Отправить свою хорошую работу в базу знаний просто. Используйте форму, расположенную ниже
Студенты, аспиранты, молодые ученые, использующие базу знаний в своей учебе и работе, будут вам очень благодарны.
Размещено на http://www.allbest.ru/
Введение
Титрование - это постепенное прибавление титрованного раствора реагента (титранта) к анализируемому раствору для определения точки эквивалентности. Титриметрический метод анализа основан на измерении объема реагента точно известной концентрации, затраченного на реакцию взаимодействия с определяемым веществом. Точка эквивалентности - момент титрования, когда достигнуто эквивалентное соотношение реагирующих веществ.
К реакциям, применяемым в количественном объемном анализе, предъявляют следующие требования:
1. Реакция должна протекать в соответствии со стехиометрическим уравнением реакции и должна быть практически необратима. Результат реакции должен отражать количество анализируемого вещества. Константа равновесия реакции должна быть достаточно велика.
2. Реакция должна протекать без побочных реакций, иначе нельзя применять закон эквивалентов.
3. Реакция должна протекать с достаточно большой скоростью, т.е. за 1-3 секунды. Это главное достоинство титриметрического анализа.
4. Должен существовать способ фиксирования точки эквивалентности. Окончание реакции должно определяться достаточно легко и просто.
Если реакция не удовлетворяет хотя бы одному из этих требований, она не может быть использована в титриметрическом анализе.
1. системы
Отличительным признаком окислительно-восстановительных реакций является перенос электронов между реагирующими частицами - ионами, атомами, молекулами и комплексами, в результате чего изменяется степень окисления этих частиц, например
Поскольку электроны не могут накапливаться в растворе, одновременно должны проходить два процесса - потери и приобретения, т. е. процесс окисления одних и восстановления других частиц. Таким образом, любая окислительно-восстановительная реакция всегда может быть представлена в виде двух полуреакций:
аOx1 + bRed2 = аRed1 + bOx2
Исходная частица и продукт каждой полуреакции составляют окислительно-восстановительную пару или систему. В вышеприведенных полуреакциях Red1 является сопряженым с Ox1, а Ox2 сопряжен с Red1.
Потенциал любой окислительно-восстановительной системы, измеренный в стандартных условиях относительно водородного электрода, называют стандартным потенциалом (Е0) этой системы. Стандартный потенциал принято считать положительным, если система выступает в качестве окислителя и на водородном электроде протекает полуреакция окисления:
или отрицательным, если система играет роль восстановителя, а на водородном электроде происходит полуреакция восстановления:
Абсолютное значение стандартного потенциала характеризует «силу» окислителя или восстановителя.
Стандартный потенциал - термодинамическая стандартизированная величина - является очень важным физико-химическим и аналитическим параметром, позволяющим оценивать направление соответствующей реакции и рассчитывать активности реагирующих частиц в условиях равновесия.
Для характеристики окислительно-восстановительной системы в конкретных условиях пользуются понятием реального (формального) потенциала Е0", который соответствует потенциалу, установившемуся на электроде в данном конкретном растворе при равенстве 1 моль/л исходных концентраций окисленной и восстановленной форм потенциалопределяющих ионов и зафиксированной концентрации всех прочих компонентов раствора.
Реальные потенциалы с аналитической точки зрения более ценны, чем стандартные потенциалы, так как истинное поведение системы определяется не стандартным, а реальным потенциалом и именно последний позволяет предвидеть протекание окислительно-восстановительной реакции в конкретных условиях. Реальный потенциал системы зависит от кислотности, присутствия посторонних ионов в растворе и может изменяться в широком диапазоне.
2. Кривые титрования
В титриметрических методах расчет и построение кривой титрования дают возможность оценить, насколько успешным будет титрование, и позволяют выбрать индикатор. При построении кривой окислительно-восстановительного титрования по оси ординат откладывают потенциал системы, а по оси абсцисс - объем титранта или процент оттитровывания.
2.1 Влияние условий титрования на ход кривых
Кривая титрования построена, исходя из значений окислительно-восстановительных потенциалов, поэтому все факторы, влияющие на потенциал, будут оказывать влияние на форму кривой титрования и скачок на ней. К таким факторам относят значения стандартного потенциала систем определяемого вещества и титранта, число электронов, участвующих в полуреакциях, рН раствора, присутствие комплексообразующих реагентов или осадителей, природу кислоты. Чем большее число электронов принимает участие в окислительно-восстановительной реакции, тем более пологая кривая характеризует данное титрование. Скачок титрования тем больше, чем больше разница окислительно-восстановительных потенциалов окислителя и восстановителя. При очень малой разнице их окислительно-восстановительных потенциалов титрование невозможно. Так титрование ионов Cl- (Е = 1,36В) перманганатом (Е = 1,51) практически невозможно. Часто бывает необходимо расширить интервал потенциалов, в котором находится скачок, если он мал. В таких случаях прибегают к регулированию скачка.
Значительно влияет на размер скачка уменьшение концентрации одного из компонентов окислительно-восстановительной пары (например, с помощью комплексообразующего реагента). Предположим, что в раствор вводят фосфорную кислоту, фториды или оксалаты, образующие комплексы с железом (III) и не взаимодействующие с железом (II), при этом потенциал пары Fe3+/Fe2+ понижается. Если, например, вследствие реакции конкурирующего комплексообразования концентрация ионов Fe3+ в растворе понизится в 10 000 раз, скачок потенциала на кривой титрования начнется уже не при Е = 0,95В, а при Е = 0,71В. Окончится он, как и раньше, при Е = 1,48В. Таким образом, область скачка на кривой титрования окажется значительно расширенной.
Повышение температуры, соответственно, увеличивает потенциал системы титранта и определяемого вещества.
Итак, при выборе оптимальных условий окислительно-восстановительного титрования следует прежде всего учитывать их влияние на состояние окислительно-восстановительной системы, а следовательно, на реальный окислительно-восстановительный потенциал.
2.2 Определение точки эквивалентности
В окислительно-восстановительных методах титрования, так же как и в методах кислотно-основного взаимодействия, возможны различные способы индикации точки эквивалентности.
1. Безындикаторные методы применимы при использовании окрашенных титрантов (растворы KMnO4, I2), незначительный избыток которых придает раствору визуально фиксируемую окраску.
2. Индикаторные методы могут быть химическими, если при этом используют в качестве индикаторов химические соединения, резко изменяющие свою окраску вблизи точки эквивалентности (в пределах скачка на кривой титрования).
Иногда в окислительно-восстановительных методах титрования применяют кислотно-основные индикаторы: метиловый оранжевый, метиловый красный, конго красный и др. Эти индикаторы в конечной точке титрования необратимо окисляются избытком окислителя и при этом меняют свою окраску.
Возможно применение флуоресцентных и хемилюминесцентных индикаторов при титровании восстановителей сильными окислителями. К числу флуоресцентных индикаторов относят многие вещества (акридин, эухризин и др.), излучающие в видимой области при определенных значениях рН раствора после облучения их ультрафиолетовым излучением. Хемилюминесцентными индикаторами являются вещества (люминол, люцигенин, силоксен и др.), излучающие в видимой области спектра в конечной точке титрования вследствие экзотермических химических процессов. Хемилюминесценция наблюдается главным образом при реакциях окисления пероксидом водорода, гипохлоритами и некоторыми другими окислителями. Достоинством флуоресцентных и хемилюминесцентных индикаторов является то, что их можно применять для титрования не только прозрачных и бесцветных, но и мутных или окрашенных растворов, для титрования которых обычные редокс-индикаторы непригодны.
Индикаторные методы могут быть также физико-химическими: потенциометрические, амперометрические, кондуктометрические и др.
2.3 Окислительно-восстановительные индикаторы
Для определения точки эквивалентности в редоксиметрии используют различные индикаторы:
1. Окислительно-восстановительные индикаторы (редокс-индикаторы), изменяющие цвет при изменении окислительно-восстановительного потенциала системы.
2. Специфические индикаторы, изменяющие свой цвет при появлении избытка титранта или исчезновении определяемого вещества. Специфические индикаторы применяют в некоторых случаях. Так крахмал - индикатор на присутствие свободного йода, вернее трииодид-ионов. В присутствии крахмал при комнатной температуре синеет. Появление синей окраски крахмала связано с адсорбцией на амилазе, входящей в состав крахмала.
Иногда в качестве индикатора используют тиоцианат аммония при титровании солей железа(III), катионы с ионами образуют соединение красного цвета. В точке эквивалентности все ионы восстанавливаются до и титруемый раствор из красного становится бесцветным.
При титровании раствором перманганата калия сам титрант играет роль индикатора. При малейшем избытке KMnO4 раствор окрашивается в розовый цвет.
Редокс-индикаторы делятся на: обратимые и необратимые.
Обратимые индикаторы - обратимо изменяют свой цвет при изменении потенциала системы. Необратимые индикаторы - подвергаются необратимому окислению или восстановлению, в результате чего цвет индикатора изменяется необратимо.
Редокс-индикаторы существуют в двух формах окисленной и восстановленной, причем цвет одной формы отличается от цвета другой.
Переход индикатора из одной формы в другую и изменение его окраски происходит при определенном потенциале системы (потенциале перехода). Потенциал индикатора определяется по уравнению Нернста:
При равенстве концентраций окисленной и восстановленной форм индикатора. При этом половина молекул индикатора существует в окисленной форме, половина - в восстановленной форме. Интервал перехода индикатора (ИП) лежит в пределах отношений концентраций обеих форм индикатора от 1/10 до 10/1.
При проведении окислительно-восстановительного титрования необходимо подбирать индикатор таким образом, чтобы потенциал индикатора находился в пределах скачка потенциала на кривой титрования. Многие индикаторы окислительно-восстановительного титрования обладают кислотными или основными свойствами и могут менять свое поведение в зависимости от рН среды.
Одним из наиболее известных и употребимых редокс-индикаторов является дифениламин:
Восстановленная форма индикатора бесцветная. Под действием окислителей дифениламин сначала необратимо переходит в бесцветный дифенилбензидин, который затем обратимо окисляется до сине-фиолетового дифенилбензидинфиолетового.
Двухцветным индикатором является ферроин, представляющий собой комплекс Fe2+ с о-фенантролином
Титрование индикаторным методом возможно, если для данной реакции ЭДС? 0,4В. При ЭДС = 0,4-0,2В используют инструментальные индикаторы.
3. Классификация методов окислительно-восстановительного титрования
Если окислительно-восстановительная реакция протекает нестехеометрично или недостаточно быстро, применяют косвенные способы титрования: обратное титрование и титрование по замещению. Например, при цериметрическом определении Fe3+ используют способ титрования по замещению:
Fe3+ +Ti3+ = TiIV + Fe2+ + + CeIV = Fe3+ + Ce3+.3+ не мешает титрованию.
Окислительно-восстановительное титрование возможно, если в растворе присутствует одна подходящая степень окисления определяемого компонента. В противном случае до начала титрования необходимо провести предварительное восстановление (окисление) до подходящей степени окисления, как это делают, например, при анализе смеси Fe2+ и Fe3+ методом перманганатометрии. Предварительное восстановление (окисление) должно обеспечить количественный перевод определяемого элемента в нужную степень окисления.
Вводимый для этой цели реагент должен представлять собой такое соединение, от избытка которого перед началом титрования легко освободиться (кипячением, фильтрованием и др.). В некоторых случаях методом редоксиметрии определяют соединения, не изменяющие своей степени окисления.
Так, титрованием по замещению, определяют ионы кальция, цинка, никеля, кобальта и свинца в перманганатометрии, сильные кислоты - в иодометрии.
Таблица 1
Методы окислительно-восстановительного титрования
|
Название метода |
Стандартный раствор (титрант) |
Уравнения полуреакций системы титранта |
Особенности метода |
||
|
Стандартный раствор - окислитель |
|||||
|
Перманганато-метрия |
MnO4?+ 8H+ + 5e? = Mn2++ 4H2O MnO4?+ 4H+ + 3e? = MnO2 + 2H2O MnO4?+ 2H2O + 3e? = MnO2+ 4OH? |
Безындикаторный метод, используется в широкой области рН |
|||
|
Броматометрия |
BrO3?+ 6H+ + 6e? = Br?+ 3H2O |
Индикатор - мети-ловый оранжевый. Среда - сильнокислая |
|||
|
Цериметрия |
Ce4+ + e? = Ce3+ |
Индикатор - ферроин. Среда - сильнокислая |
|||
|
Хроматометрия |
Сr2O72?+ 14H+ + 6e? = 2Cr3++2H2O |
Индикатор - дифе-ниламин. Среда? сильнокислая |
|||
|
Нитритометрия |
NO2- + 2H+ + e? = NO + H2O |
Внешний индикатор - иодид- крахмаль-ная бумага. Среда? слабокислая |
|||
|
Иодиметрия |
Индикатор - крахмал |
||||
|
Стандартный раствор - восстановитель |
|||||
|
Аскорбино-метрия |
С6H6O6 +2H+ +2 e? = С6H8O6 |
Индикаторы - вари-аминовый синий или для определе-ния ионов Fe3+ роданид калия. Среда - кислая |
|||
|
Титанометрия |
TiO2+ + 2H+ + e? =Ti3+ + H2O |
Индикатор - мети-леновый голубой. Среда - кислая |
|||
|
Иодометрия |
S4O62?+ 2e? = 2S2O32? |
Индикатор - крах-мал. Вспомогатель-ный реагент - KI. Среда - слабокислая или нейтральная |
4. Перманганатометрия
Перманганатометрия - один из наиболее часто применяемых методов окислительно-восстановительного титрования. В качестве титранта используют раствор перманганата калия, окислительные свойства которого можно регулировать в зависимости от кислотности раствора.
4.1 Особенности метода
Наибольшее распространение в аналитической практике получил перманганатометрический метод определения в кислых средах: восстановление MnO4- до Mn2+ проходит быстро и стехиометрично:
Особенностью метода является сильное влияние концентрации ионов водорода на стандартный потенциал системы MnO4-/ Mn2+. При титровании в сильнокислых средах чаще всего используют серную кислоту. Хлороводородную и азотную кислоты применять не следует, так как в их присутствии могут идти конкурирующие окислительно-восстановительные реакции. Восстановление перманганат-иона в щелочной среде протекает последовательно: сначала до манганат-иона MnO42-, а затем до диоксида марганца MnO2:
Количественно восстановление перманганата в щелочной среде до манганата протекает в присутствии соли бария. Ba(MnO4)2 растворим в воде, в то время как ВаMnO4 - нерастворим, поэтому дальнейшее восстановление MnVI из осадка не происходит.
Перманганатометрически в щелочной среде, как правило, определяют органические соединения: формиат, формальдегид, муравьиную, коричную, винную, лимонную кислоты, гидразин, ацетон и др.
Индикатором конца титрования служит бледно-розовая окраска избытка титранта КMnO4 (одна капля 0,004 М раствора титранта придает заметную окраску 100 мл раствора). Поэтому, если титруемый раствор бесцветен, о достижении точки эквивалентности можно судить по появлению бледно-розовой окраски избытка титранта КMnO4 при титровании прямым способом или по исчезновению окраски при реверсивном титровании. При анализе окрашенных растворов рекомендуется использовать индикатор ферроин.
К достоинствам перманганатометрического метода относят:
1. Возможность титрования раствором КMnO4 в любой среде (кислой, нейтральной, щелочной).
2. Применимость раствора перманганата калия в кислой среде для определения многих веществ, которые не взаимодействуют с более слабыми окислителями.
Наряду с перечисленными достоинствами метод перманганатометрии имеет ряд недостатков:
1. Т итрант КMnO4 готовят как вторичный стандарт, поскольку исходный реагент - перманганат калия - трудно получить в химически чистом состоянии.
2. Реакции с участием MnO4- возможны в строго определенных условиях (рН, температура и т. д.).
4.2 Применение метода
1. Определение восстановителей. Если окислительно-восстановительная реакция между определяемым восстановителем и MnO4- протекает быстро, то титрование проводят прямым способом. Так определяют оксалаты, нитриты, пероксид водорода, железо (II), ферроцианиды, мышьяковистую кислоту и др.:
Н2О2 + 2MnO4- + 6Н+ = 5О2 + 2Мn2+ + 8Н2О
54- + MnO4- + 8H+ = 53- + 2Mn2+ + 4H2O
AsIII + 2MnO4- + 16H+ = 5AsV + 2 Mn2+ + 8H2O
5Fe2+ + MnO4- +8H+ = 5Fe3+ + 2Мn2+ + 4Н2О
2. Определение окислителей. Добавляют избыток стандартного раствора восстановителя и затем титруют его остаток раствором KMnO4 (способ обратного титрования). Например, хроматы, персульфаты, хлориты, хлораты и другие окислители можно определять перманганатометрическим методом, подействовав сначала избытком стандартного раствора Fe2+, а затем оттитровав непрореагировавшее количество Fe2+ раствором KMnO4:
Cr2O72- + 6Fe2+ + 14H+ = 2Cr3+ + 6Fe3+ + 7H2O + (Fe2+) - избыток-
Fe2+ + MnO4- + 8H+ = 5Fe3+ + Mn2+ + 4H2O - остаток
3. Определение веществ, не обладающих окислительно-восстановительными свойствами, проводят косвенным способом, например титрованием по замещению. Для этого определяемый компонент переводят в форму соединения, обладающего восстановительными или окислительными свойствами, а затем проводят титрование. Например, ионы кальция, цинка, кадмия, никеля, кобальта, осаждают в виде малорастворимых оксалатов:
М2+ + С2О4- = vМС2О4
Осадок отделяют от раствора, промывают и растворяют в H2SO4:
МС2О4 + H2SO4 = H2C2O4 + MSO4
Затем H2C2O4 (заместитель) титруют раствором KMnO4:
2MnO4- + 5С2O42- + 16H+ = 2Mn2+ +10CO2 + 8H2O
4. Определение органических соединений. Отличительной особенностью реакций органических соединений с MnO4- является их малая скорость. Определение возможно, если использовать косвенный способ: анализируемое соединение предварительно обрабатывают избытком сильнощелочного раствора перманганта и дают возможность реакции протекать необходимый период времени. Остаток перманганата титруют раствором оксалата натрия:
С3Н5(ОН)3 + 14MnO4- + 20OH- = 3CO32- + 14MnO42- + 14H2O +
(MnO4-), избыток остаток
2MnO4- + 5С2O42- + 16H+ = 2Mn2+ +10CO2 + 8H2O остаток
окислительный восстановительный титриметрический
5. Суть и классификация осадительных методов
Методы осадительного титрования - это методы титриметрического анализа, в которых применяются титранты, которые образуют осадки с определяемыми веществами.
Требования к реакциям и определяемым веществам:
1. Определяемое вещество должна быть хорошо растворимо в воде и должно образовывать ионы, которые были бы активными в реакциях осаждения.
2. Получаемый в реакции осадок должен быть практически нерастворимым (ПР < 10 -8 ? - 10 , S < 10 -5).
3. Результаты титрования не должны искажаться явлениями адсорбции (соосаждение).
4. Выпадание осадка должно происходить достаточно быстро (т.е. не должны образовываться пересыщенные растворы).
5. Должна быть возможность фиксации точки эквивалентности.
Классификация методов осадительного титрования в зависимости от используемых титрантов:
Аргентометрия (титрант AgNO 3) ;
Меркурометрия (титрант Hg 2 (NO 3) 2);
Тиоцианатометрия (титрант NH 4 SCN);
Сульфатометрия (титранты H 2 SO 4 , BaCl 2);
Хроматометрия (титрант K 2 CrO 4);
Гексацианоферратометрия (титрант K 4 ).
6. Кривые титрования и их анализ
Построение кривых титрования осуществляется на основании расчетов согласно правилу произведения растворимости и соответственно.
Кривая титрования строится в координатах, которые показывают изменение концентрации определяемого иона в зависимости от объема добавленного титранта.
Чем больше скачок титрования на кривой, тем более широкие возможности для выбора соответствующего индикатора.
Факторы, которые влияют на величину скачка на кривых осадительного титрования:
1. Концентрация растворов титранта и определяемого иона Чем выше концентрация, тем больше скачок на кривой титрования.
2. Растворимость осадка, который образуется в процессе титрования (чем меньше растворимость, тем больше скачок титрования).
Зависимость величины скачка титрования от растворимости труднорастворимого электролита.
3. Температура
Чем выше температура, тем больше растворимость осадка и тем меньше скачок на кривой титрования. Титрование проводят при комнатной температуре.
4. Ионная сила раствора
Влияние относительно незначительное, так как ионная сила раствора, по сравнению с другими факторами, не так сильно изменяет растворимость осадка; тем не менее, чем выше ионная сила раствора, тем выше растворимость и меньше скачок титрования.
7. Аргентометрия
Аргентометрия - метод осадительного титрования, который базируется на реакциях образования трудно растворимых солей Аргентума:
X - + Ag + = AgХ,
где X - = Cl - , Br - , I - , CN - , SCN - и др.
Титрант: AgNO 3 - вторичный стандартный раствор.
Стандартизация: за первичным стандартным раствором натрий хлорида NaCl:
Индикатором при стандартизации есть 5 % калий хромат K 2 CrО 4 . Титрование проводят до появления коричнево-красного осадка аргентум хромата:
В зависимости от способа проведения титрования и используемого индикатора методы аргентометрии классифицируют на:
Безиндикаторные: - метод Гей-Люссака (метод равного помутнения)
Метод до точки просветления
Индикаторные: - метод Мора
Метод Фаянса - Фишера - Ходакова
Метод Фольгарда
Метод Мора
Титрант: AgNO 3 - втор. станд. раствор.
AgNO 3 + NaCl = AgCl? + NaNO 3
Индикатором есть 5% калий хромат K 2 CrО 4 (до появления коричнево-красного аргентум хромата):
2AgNO 3 + K 2 CrО 4 = Ag 2 CrО 4 ?+ 2KNO 3
Определяемые вещества: хлориды Cl - , бромиды Br - .
Среда: рН~ 6,5-10,3.
Применение: количественное определение натрий хлорида, калий хлорида, натрий бромида, калий бромида в субстанция лекарственных веществ.
Ограничения применения:
1. Нельзя титровать кислые растворы:
2CrО 4 2- + 2H + = Cr 2 O 7 2- + H 2 O
2. Нельзя титровать в присутствии аммиака и других ионов, молекул, которые могут выступать лигандами по отношению к ионам аргентума в реакциях комплексообразования.
3. Нельзя титровать в присутствии многих катионов (Ba 2+ , Pb 2+ , и др.), которые образуют окрашенные осадки с хромат - ионами CrО 4 2- .
4. Нельзя титровать в присутствии восстановителей, которые реагируют с хромат-ионами CrО 4 2- , превращая их в ионы Cr 3+ .
5. Нельзя титровать в присутствии многих анионов (PO 4 3- , AsО 4 3- , AsО 3 3- , S 2- и др.), которые с ионами аргентума образуют окрашенные осадки аргентума.
Метод Фаянса-Фишера-Ходакова
Титрант: AgNO 3 - втор. станд. раствор
Стандартизация за перв. станд. раствором натрий хлорида NaCl методом пипетирования:
AgNO 3 + NaCl = AgCl? + NaNO 3
Индикатором при стандартизации есть 5% раствор калий хромата K 2 CrО 4 (до появления коричнево-красного осадка аргентум хромата):
2AgNO 3 + K 2 CrО 4 = Ag 2 CrО 4 ?+ 2KNO 3
Среда: рН~ 6,5-10,3 при определении хлоридов и рН~ 2,0-10,3 при определении бромидов и йодидов.
Индикаторы метода:
Флуоресцеин при определении хлоридов;
Эозин при определении бромидов и йодидов.
Механизм действия индикаторов: адсорбционный. Адсорбционные индикаторы - это индикаторы, адсорбция или десорбция которых осадком сопровождается изменением окраски в Т.Э. или вблизи нее.
AgNO 3 + NaCl = AgCl? + NaNO 3
HInd х H + + Ind - .
Условия проведения титрования:
1. Кислотность растворов
2. Концентрация реагирующих растворов
3. Учет адсорбционной способности индикаторов и присутствующих в растворе ионов.
4. Титрование вблизи т.э. следует проводить медленно
5. Титрование с адсорбционными индикаторами проводят в рассеянном свете.
Применение: количественное определение хлоридов, бромидов, йодидов, тиоцианатов, цианидов.
Метод Фольгарда
Титранты: AgNO 3 , аммоний или калий тиоцианат NH 4 SCN, KSCN - вторичные стандартные растворы.
Стандартизация AgNO 3 за перв. станд. раствором NaCl методом пипетирования:
AgNO 3 + NaCl = AgCl? + NaNO 3
Индикатором при стандартизации AgNO 3 есть 5 % раствор калий хромата K 2 CrО 4 (до появления коричнево-красного осадка аргентум хромата):
2AgNO 3 + K 2 CrО 4 = Ag 2 CrО 4 + 2KNO 3
Стандартизация NH 4 SCN, KSCN за стандартным раствором AgNO 3:
AgNO 3 + NH 4 SCN = AgSCN + NH 4 NO 3
Индикатором при стандартизации аммоний или калий тиоцианата являются соли ферума (ІІІ) (например, NH 4 Fe(SO 4) 2 12H 2 O в присутствии нитратной кислоты):
Fe 3+ + SCN - = 2+
Титруют до появления слабо розовой окраски.
Среда: нитратнокислая.
Индикаторы метода: соли ферума (ІІІ) NH 4 Fe(SO 4) 2 ?12H 2 O в присутствии нитратной кислоты.
Определяемые вещества: галогенид-ионы, цианиды, тиоцианаты, сульфиды, карбонаты, хроматы, оксалаты, арсенаты и др.
Hal - + Ag + (избыток) = AgHal
Ag + (остаток) + SCN - = AgSCN,
а после точки эквивалентности:
Fe 3+ + SCN - = 2+
(розово-красная окраска)
При определении йодидов индикатор прибавляют в конце титрования, во избежание параллельной реакции:
2Fe 3+ + 2I - = 2Fe 2+ + I 2
Преимущества метода Фольгарда - возможность титрования:
В очень кислых растворах;
В присутствии многих катионов, которые мешали при определении по методу Мора (катионы бария, плюмбума и др., которые образовывали окрашенные осадки хроматов).
8. Меркурометрия
Меркурометрия - это метод осадительного титрования, который базируется на использовании реакций образования труднорастворимых осадком солей меркурия (І) Hg 2 2+ :
2Cl - + Hg 2 2+ = Hg 2 Cl 2 Ї ПР = 1,3Ч10 -18
2I - + Hg 2 2+ = Hg 2 I 2 Ї ПР = 4,5 Ч10 -29
Титрант: втор. станд. раствор Hg 2 (NO 3) 2 .
Стандартизация: за стандартным раствором NaCl:
Hg 2 (NO 3) 2 + 2NaCl = Hg 2 Cl 2 Ї + 2NaNO 3
Индикаторы: 1) раствор ферум (ІІІ) тиоцианата (от красного до обесцвечивания)
2Fe(SCN) 2+ + Hg 2 2+ = Hg 2 (SCN) 2 Ї + 2Fe 3+ ;
1-2% спиртовый раствор дифенилкарбазона (до появления синей окраски).
Для учета объема титранта, который израсходовано на титрование индикатора титруют “слепую пробу”:
2) Индикатор прибавляют перед окончанием титрования, так как если его прибавить сначала, то может задолго до т.э. образоваться дифенилкарбазид меркурия (ІІ) и дать синюю окраску скорее, чем будет оттитрован галогенид.
Определяемые вещества: хлориды и йодиды.
Среда: очень кислая (может быть до 5 моль/л ионов H +).
Недостаток: соли Меркурия (І) - очень токсичны.
9. Сульфатометрия
Сульфатометрия - метод осадительного титрования, который базируется на использовании реакций образования труднорастворимых солей - сульфатов.
Иногда выделяют бариметрию - метод осадительного титрования, который базируется на использовании реакций образования труднорасторимых солей бария.
В основе метода лежит реакция образования осадка барий сульфата:
Ba 2+ + SO 4 2- = BaSO 4 Ї
определ. вещество титрант
Титранты: втор. станд. растворы H 2 SO 4 , Ba(NO 3) 2 , BaCl 2 .
Стандартизация: раствор H 2 SO 4 по Na 2 B 4 O 7 или Na 2 CO 3 с метиловым оранжевым; Ba(NO 3) 2 и BaCl 2 по H 2 SO 4 с нитрхромазо или ортаниловым А.
Индикаторы: применяют металлохромные индикаторы (изменяют свою окраску в присутствии ионов металлов) - нитрхромазо (ортаниловый С), ортаниловый А. Эти индикаторы в растворе окрашены в розовый цвет, а в присутствии катионов бария в фиолетовый цвет.
Определяемые вещества в прямом титровании:
сульфатной кислотой - содержание бария;
барий хлоридом или барий нитратом - содержание сульфатов.
Заключение
Из титриметрических методов анализа окислительно-восстановительное титрование является широко распространенным, границы применения этого метода шире, чем кислотно-основного или комплексонометрического методов. Благодаря большому разнообразию окислительно-восстановительных реакций этот метод позволяет определять большое количество самых разнообразных веществ, в том числе и тех, которые непосредственно не проявляют окислительно-восстановительных свойств.
Перманганатометрия используется для определения общей окисляемости воды и почвы. При этом с MnO4--ионом в кислой среде реагируют все органические компоненты (в том числе гуминовые кислоты почв и природных вод). Число миллимоль эквивалентов KMnO4, пошедших на титрование, и является характеристикой окисляемости (по перманганату).
Перманганатометрию применяют и для анализа легко окисляющихся органических соединений (альдегидов, кетонов, спиртов, карбоновых кислот: щавелевой, винной, лимонной, яблочной, а также гидразогрупп). В пищевой промышленности перманганатометрию можно использовать для определения содержания сахара в пищевых продуктах и сырье, содержания нитритов в колбасных изделиях.
В металлургической промышленности методом перманганатометрии определяют содержание железа в солях, сплавах, металлах, рудах и силикатах.
Список литературы
1. Аналитическая химия. Химические методы анализа/ под ред. О.М. Петрухина. М.: Химия, 1992, 400 с.
2. Васильев В.П. Аналитическая химия. В 2 ч. Ч. 1. Гравиметрический и титриметрический методы анализа. М.: Высшая школа, 1989, 320 с.
3. Основы аналитической химии. В 2 кн. Кн. 2. Методы химического анализа/ под ред. Ю.А. Золотова. М.: Высшая школа, 2000, 494 с.
Размещено на Allbest.ru
...Подобные документы
Отличительные признаки окислительно-восстановительных реакций. Схема стандартного водородного электрода. Уравнение Нернста. Теоретические кривые титрования. Определение точки эквивалентности. Окислительно-восстановительные индикаторы, перманганатометрия.
курсовая работа , добавлен 06.05.2011
Классификация методов окислительно-восстановительного титрования. Индикаторы окислительно-восстановительного титрования. Перманганатометрия, йодометрия и дихроматометрия. Окраска окисленной и восстановленной формы. Фиксирование точки эквивалентности.
реферат , добавлен 23.02.2011
Особенности методов окислительно-восстановительного титрования. Основные требования к реакциям, константа равновесия. Характеристика видов окислительно-восстановительного титрования, его индикаторы и кривые. Приготовление и стандартизация растворов.
курсовая работа , добавлен 25.12.2014
Классификация методов титраметрического анализа. Сущность метода "нейтрализации". Приготовление рабочих растворов. Расчет точек и построение кривых кислотно-основного и окислительно-восстановительного титрования. Достоинства и недостатки йодометрии.
курсовая работа , добавлен 17.11.2013
Классификация методов окислительно-восстановительного титрования. Факторы, оказывающие влияние на скорость реакции. Специфические и редокс-индикаторы. Сущность перманганатометрии, иодометрии, дихроматометрии. Приготовление раствора дихромата калия.
презентация , добавлен 19.03.2015
Метод кислотно-основного титрования: понятие и содержание, основные этапы и принципы реализации, предъявляемые требования, главные условия и возможности применения. Расчет рН растворов. Построение кривых титрования. Выбор индикатора и его обоснование.
презентация , добавлен 16.05.2014
Понятие титраметрического анализа. Окислительно-восстановительное титрование, его виды и условия проведения реакций. Расчет точек кривой титрования, потенциалов, построение кривой титрования. Подборка индикатора, расчет индикаторных ошибок титрования.
курсовая работа , добавлен 10.06.2012
Титриметрический метод анализа. Теория броматометрического метода анализа. Техника титрования. Достоинства и недостатки броматометрического метода. Фенолы. Определение фенола. Химические реакции, используемые в методах титриметрии.
курсовая работа , добавлен 26.03.2007
Классификация окислительно-восстановительного титрования; его применение в фармацевтическом анализе, при определении окисляемости воды и органических соединений. Рассмотрение редокс-титрования на примере цериметрии. Титрование соли железа сульфатом церия.
курсовая работа , добавлен 12.09.2012
Определение кристаллизационной воды в хлориде бария. Установка титра рабочего раствора соляной кислоты. Метод кислотно-основного и окислительно-восстановительного титрования. Определение содержания ионов в растворе методом качественного анализа.
Титриметрический анализ
История и принцип метода
Титриметрический анализ (титриметрия) -важнейший из химических методов анализа. Он возник в XVIII веке, вначале как эмпирический способ проверки качества различных материалов, например, уксуса, соды, отбеливающих растворов. На рубеже XVIII и XIX веков были изобретены бюретки и пипетки (Ф.Декруазиль). Особое значение имели труды Ж.Гей-Люссака, который ввел основные термины этого метода: титрование, титрант и другие, происходящие от слова «титр». Титр – это массарастворенного вещества (в граммах), содержащаяся в одном миллилитре раствора. Во времена Гей-Люссака результаты анализа вычисляли именно с помощью титров. Однако титр как способ выражения концентрации раствора оказался менее удобным, чем другие характеристики (например, молярные концентрации), поэтому в современной аналитике химии расчеты с применением титров ведут довольно редко. Напротив, различные термины, произведенные от слова «титр», применяют очень широко.
В середине XIX века немецкий химик К.Мор обобщил все созданные к тому времени титриметрические методики и показал, что в основе любой методики лежит один и тот же принцип. К раствору пробы, содержащей определяемый компонент Х, всегда прибавляют раствор с точно известной концентрацией реагента R (титрант). Этот процесс и называют титрованием. Проводя титрование, аналитик следит за протеканием химической реакции между Х и добавляемым R . По достижении точки эквивалентности (т.экв.), когда число молей эквивалентов введенного R точно сравняется с числом молей эквивалентов находившегося в пробе вещества Х, титрование прекращают и измеряютобъем затраченного титранта. Момент окончания титрования называют конечной точкой титрования (к.т.т.), ее, как и т.экв., выражают в единицах объема, обычно в миллилитрах. В идеальном случае V к.т.т = V т.экв. , но на практике точное совпадение по разным причинам не достигается, титрование заканчивают чуть раньше или, наоборот, чуть позже, чем будет достигнута т.экв. Естественно, титрование следует проводить так, чтобы различие между V т.экв. и V к.т.т. было бы как можно меньшим.
Поскольку массу или концентрацию Х рассчитывают по объему титранта, затраченному на титрование пробы (по V к.т.т.), впрошлом титриметрию называли объемным анализом . Это название нередко используют и сегодня, но термин титриметрический анализ более точен. Дело в том, что операция постепенного прибавления реагента (титрование) характерна для любой методики этого типа, а расход титранта можно оценивать не только путем измерения объема, но и другими способами. Иногда добавляемый титрант взвешивают (измерение массы на аналитических весах дает меньшую относительную погрешность, чем измерение объема). Иногда измеряют время, за которое будет введен титрант (при постоянной скорости ввода).
С конца XIX века титриметрические методики стали применять и в исследовательских,и в заводских, и в других лабораториях. С помощью нового метода оказалось возможным определять миллиграммовые и даже микрограммовые количества самых разных веществ. Широкому использованию титриметрии способствовали простота метода, невысокая стоимость и универсальность оборудования. Особенно широко титриметрию стали применять в 50-х годах XX века,после создания швейцарским аналитиком Г.Шварценбахомнового варианта этого метода (комплексонометрии). Одновременно началось широкое применение инструментальных методов контроля к.т.т. К концу 20 века значение титриметрии несколько снизилось в связи с конкуренцией более чувствительных инструментальных методов, но и сегодня титриметрия остается очень важным методом анализа. Она позволяет быстро, легко и достаточноточно определять содержание большинства химических элементов, отдельные органические и неорганические вещества, суммарное содержание однотипных веществ, а также обобщенные показатели состава (жесткость воды, жирность молока, кислотность нефтепродуктов).
Техника проведения титриметрического анализа
Принцип метода станет более понятен после изложения техники его проведения. Итак, пустьВам принесли раствор щелочи неизвестной концентрации, и Ваша задача – установить его точную концентрацию. Для этого Вам понадобится раствор регента , или титранта – вещества, которое вступает в химическую реакцию со щелочью, причем концентрация титранта должна быть точно известна. Очевидно, что для установления концентрации щелочи в качестве титранта используемраствор кислоты.
1. Отбираем с помощью пипетки точный объем анализируемого раствора – он называется аликвота . Как правило, объем аликвоты составляет 10-25 мл.
2. Переносим аликвоту в колбу для титрования, разбавляем водой и добавляем индикатор.
3. Заполняем бюретку раствором титранта и выполняем тирование – медленное, по каплям, добавление титранта к аликвоте исследуемого раствора.
4. Заканчиваем титрование в момент, когда индикатор изменит свою окраску. Этот момент называется конечной точкой титрования – к.т.т. К.т.т., как правило, совпадает с моментом, когда реакция между определяемым веществом и титрантом закончена, т.е. к аликвоте добавлено точно эквивалентное количество титранта – этот момент называется точкой эквивалентности, т.э. Таким образом т.э. и к.т.т. – это две характеристики одного и того же момента, одна – теоретическая, другая – экспериментальная, зависящая от выбранного индикатора. Поэтому надо правильно выбирать индикатор, с тем, чтобы к.т.т. как можно точнее совпадала с т.э.
5. Измеряют объем титранта, пошедшего на титрование, и вычисляют концентрацию исследуемого раствора.
Виды титриметрического анализа
Классифицировать титриметрические методики можно по нескольким независимым признакам: а именно: 1) по типу реакции между Х и R , 2) по способу проведения титрования и расчета результатов,3) по способу контроля т.экв.
Классификация по типу химической реакции – наиболее важная. Напомним, что далеко не все химические реакции можно использовать для проведения титрований.
Во-первых, как и в других химических методах, определяемый компонент (аналит) должен количественно реагировать с титрантом.
Во-вторых, надо, чтобы равновесие реакции устанавливалось как можно быстрее. Реакции, в которых после добавления очередной порции титранта установление равновесия требует хотя бы нескольких минут, в титриметрии применять затруднительно или вообще невозможно.
В-третьих, реакция должна отвечать единственному и заранее известному стехиометрическому уравнению. Если реакция ведет к смеси продуктов, состав этой смеси будет меняться в ходе титрования и зависеть от условий проведения реакции. Зафиксировать точку эквивалентности будет очень трудно, а результат анализа окажетсянеточным.Совокупности указанных требований отвечают реакции протолиза (нейтрализации), многие реакции комплексообразования и окисления-восстановления, а также некоторые реакции осаждения. Соответственно в титриметрическом анализе выделяют:
Метод нейтрализации,
Комплексометрию,
Редоксметрические методы
Методы осаждения.
Внутри каждого метода выделяют отдельные его варианты (табл.1). Их названия происходят от наименований реагентов, используемых в каждом из вариантов в качестве титранта (перманганатометрия, иодометрия, хроматометрия и т.п.).
Таблица 1.
Классификация титриметрических методик по типу используемой химической реакции
|
Реакция |
Метод |
Реагент (титрант) |
Вариантметода |
Определяемыевещества |
|
Протолиз |
Методнейтрализации |
Н Cl, HClO 4 , HNO 3 |
Ацидиметрия |
Oc нования |
|
KOH, NaOH и др. |
Алкалиметрия |
Кислоты |
||
|
Комплексо-образование |
Комплексо-метрия |
ЭДТА |
Комплексонометрия |
Металлы и ихсоединения |
|
Фторидометрия, цианидометрия |
Некоторые металлы, органическиевещества |
|||
|
Окисление-восстанов-ление |
Редокс-метрия |
KMnO 4 К 2 С r 2 O 7 |
Перманганатометрия хроматометрия |
Восстановители |
|
KJ и Na 2 S 2 O 3 |
Иодометрия |
Восстановители,окислители, кислоты |
||
|
Аскорбинометрия |
Окислители |
|||
|
Осаждение |
Седиметрия |
AgNO 3 |
Аргентометрия |
Галогениды |
|
Hg 2 (NO 3) 2 |
Меркуриметрия |
|||
|
KSCN |
Роданометрия |
Некоторые металлы |
||
|
Ba(NO 3) 2 |
Бариеметрия |
Сульфаты |
Классификация по способу титрования. Обычно выделяют три способа: прямое, обратное и заместительное титрование. Прямое титрование предполагает непосредственное прибавление титранта к раствору пробы. Иногда применяют другой порядок смешивания реагентов – к известному количеству R постепенно добавляют раствор пробы, в котором хотят определить концентрацию Х; но это тоже прямое титрование. В обоих случаяхрасчет результатов анализа ведут по одним и тем же формулам, основанным на законе эквивалентов.
ν Х = ν R
где ν Х иν R – количества молей эквивалентов Х и R . Расчетные формулы, основанные на соотношении, а также примеры расчетов будут даны ниже.
Прямое титрование - удобный и самый распространенный вариант титриметрии. Он более точен, чем другие. Ведь случайные погрешности в основном возникают при измерении объема растворов, а в данном способе титрования объем измеряют только один раз.Однако прямое титрование возможно далеко не всегда. Многие реакции между Х и R идут недостаточно быстро, и после добавления очередной порции титранта в растворе не успевает установиться равновесие. Иногда прямое титрование невозможно из-за побочных реакций или ввиду отсутствия подходящего индикатора. В подобных случаях применяют более сложные схемы обратного или заместительного титрования. Они включают не менее двух химических реакций.
Обратное титрование проводят по двухстадийной схеме:
Х + R 1 =Y 1
R 1 + R 2 = Y 2
Вспомогательный реагент R 1 вводят в точно известном количестве. Объем и концентрацию раствора R 1 выбирают так, чтобы R 1 после завершения реакции с Хостался в избытке. Затем непрореагировавшую часть R 1 оттитровывают титрантом R 2 . Примером может быть перманганатометрическое титрование органических веществ. Титровать многие веществаперманганатом «напрямую» не удается из-за замедленности их окисления и по другим причинам. Но можно сначала добавить к анализируемой пробе известное (избыточное) количество KMnO 4 , подкислить и нагреть полученный раствор. Это приведет к полному и быстрому завершению окисления органических веществ. Затем оттитровывают оставшийся перманганат каким-либо активным восстановителем, например, раствором SnCl 2 или FeSO 4 .
Расчет результатов обратного титрования проводят, исходяиз очевидного соотношения:
ν Х =ν R 1 - ν R 2
Поскольку объемы в данном случае измеряют два раза (сначала объем раствора реагента R 1 , затем объем титранта R 2), случайная погрешность результата анализа несколько выше, чем при прямом титровании. Особенно сильно возрастает относительная погрешность анализа при малом избытке вспомогательного реагента, когдаν R 1 ≈ν R 2 .
Классификацияпо способу контроля т.экв. Известно несколько таких способов. C амый простой - безындикаторное титрование, самый распространенный – титрование с цветными индикаторами, а самые точные и чувствительные –инструментальные варианты титриметрии.
Безындикаторное титрование основано на применении реакций, которые сопровождаются изменением видимых свойств титруемого раствора. Как правило, один из реагентов (Х или R ) имеет видимую окраску. Ход такой реакции контролируют без специальных приборов и без добавления реактивов-индикаторов. Так, бесцветные восстановители титруют в кислой среде фиолетовым раствором окислителя – перманганата калия (KMnO 4). Каждая порция добавляемого титрантабудет сразу же обесцвечиваться, превращаясь под действием восстановителя в ионы Mn 2+ . Так будет продолжаться вплоть до т.экв. Однако первая же «лишняя» капля титранта окрасит титруемый раствор врозово-фиолетовый цвет, окраска не исчезнет и при перемешивании раствора. При появлении неисчезающей окраски титрование прекращают иизмеряют объемзатраченного титранта (V к.т.т.). Конец титрованияможно зафиксировать не только по появлению окраски титруемого раствора, как в рассмотренном примере, но ипо обесцвечиванию ранее окрашенного раствора пробы, а также по появлению какого-либо осадка, его исчезновению или изменению внешнего вида. Безындикаторное титрование применяют довольно редко, так как лишь немногие реакции сопровождаются изменением видимых свойств раствора.
Инструментальное титрование . За протеканием реакции между Х и R можно следить не просто «на глаз» (визуально), но и с помощью приборов, измеряющих некоторое физическое свойство раствора. Варианты инструментальной титриметрии различают, смотря по тому, какое именно свойство раствора контролируется. Можно использовать любое свойство, зависящее от качественного и количественного состава титруемого раствора. А именно, можно измерять электропроводность раствора (этот вариант называют кондуктометрическим титрованием), потенциал индикаторного электрода, опущенного в титруемый раствор (потенциометрическое титрование), поглощение света титруемым раствором (фотометрическое титрование) и т.п.Прекратить титрование можно тогда, когдабудет достигнуто некоторое заранее выбранное значение измеряемого свойства. Например, титруют раствор кислоты щелочью до тех пор, пока не будет достигнуто значение рН = 7. Однако чаще поступают по-другому - выбранное свойство растворамногократно (или даже непрерывно) измеряют по мере ввода титранта, причем не только до, но и после ожидаемой т.экв.По полученным данным строят графическую зависимость измеренного свойства от объема добавленного титранта (кривую титрования ). Вблизи точки эквивалентностинаблюдается резкое изменение составаисвойств титруемого раствора, а на кривой титрования регистрируется скачок или излом. Например, скачок потенциала электрода, опущенного в раствор. Положение т.экв оценивают по положению перегиба на кривой. Такой вариант анализа более трудоемок и длителен, чем обычное титрование, но дает более точные результаты. За одно титрование удается определить по отдельности концентрации целого ряда компонентов.
Известно более десятка вариантов инструментальной титриметрии. В создании их важную роль сыграл американский аналитик И.Кольтгоф. Соответствующие методики различаются по измеряемому свойству раствора, по используемой аппаратуре и по аналитическим возможностям, но все они чувствительнее и селективнее, чеминдикаторные илибезындикаторные визуальные варианты титриметрии. Инструментальный контроль особенно важен, когда нельзя применять индикаторы, например, при анализе мутных или интенсивно окрашенных растворов, а также при определении микропримесей и при анализе смесей. Однако инструментальная титриметрия требует оснащения лаборатории специальными приборами, желательно - самопишущими или полностью автоматизированными, что не всегда экономически целесообразно. Во многих случаях достаточно точные и надежные результаты могут быть получены более простым и дешевым способом, основанным на применении индикаторов.
Использование индикаторов . К титруемой пробе можно заранее добавить небольшое количество специального реактива - индикатора . Титрование надо будет прекратить в тот момент, когда индикатор под действием введенного титранта изменит видимую окраску, это и есть конечная точка титрования. Важно, чтобы изменение окраски происходило не постепенно, ав результате добавления всего одной «лишней» капли титранта. В некоторых случаях индикатор меняет не свою окраску, арастворимость или характер свечения. Однако такие индикаторы (адсорбционные, флуоресцентные, хемилюминесцентные и др.) применяют намного реже, чемцветные индикаторы. Изменение окраски любого индикатора происходит благодаря химическому взаимодействию индикатора с титрантом, приводящему кпереходу индикаторав новую форму.Свойства индикаторов необходимо рассмотреть более детально.
Индикаторы
В аналитических лабораториях применяют несколько сот цветных индикаторов разного типа (кислотно-основные, металлохромные, адсорбционные и т.п.). Когда-то в качестве индикаторов использовались настойки, полученные из растений - из цветов фиалки или из особого вида лишайников (лакмус). Впервые такие индикаторы стал применять еще Р.Бойль. В настоящее время природные индикаторы не используют, поскольку они всегда являются смесью разных веществ, поэтому переход их окраски выражен недостаточно четко. Современные индикаторы – это специально синтезированные индивидуальные органические соединения. Как правило, индикаторами являются соединения ароматического ряда, молекулы которых содержат несколько функциональных групп (заместителей).Известно множество подобных соединений, но только некоторые из нихможно применять в качестве цветных индикаторов. Предполагаемый индикатор должен отвечать целому ряду требований:
· индикатор должен хорошо растворяться, даваярастворы, устойчивые при хранении;
· в растворе индикатор должен существовать в нескольких формах, различных по структуре молекулы. Между формами должно устанавливаться подвижноехимическое равновесие. Например, кислотная форма индикатора переходит в основную (и обратно),окисленная- ввосстановленную (и обратно); металлохромный индикатор обратимо связываетсяв комплекс с ионами металла, и т.п.;
· цветной индикатордолжен интенсивно поглощать свет в видимой области спектра. Окраска его раствора должна быть различима даже при очень низкой концентрации (10 -6 – 10 -7 моль/л). В этом случае можно будет вводить в титруемый раствор очень малые количества индикатора, что способствует получению более точных результатов анализа;
· разныеформыиндикаторадолжны быть различны по своей окраске, то есть по спектру поглощения в видимой области. В таком случае в ходе титрования будет наблюдаться контрастный цветовой переход.Например, переход окраски индикатора из розовой в изумрудно-зеленую хорошо заметенна глаз. Зафиксировать же конечную точку титрования (к.т.т.) по переходурозовой окраски воранжевую или фиолетовую гораздо труднее. Очень важно, насколько различны спектры поглощения двух форм индикатора. Если одна из форм индикатора максимально поглощает свет с длиной волны λ 1 , а другая- с длиной волны λ 2 , то разность∆λ = λ 1 - λ 2 характеризует контрастность цветового переход. Чем больше ∆λ, тем лучше воспринимается на глаз переход окраски индикатора. Для повышения визуальной контрастности цветового перехода иногда используют смеси разных индикаторов или к индикатору добавляют посторонний инертный краситель;
· переход индикатора из одной формы в другую при изменении состава раствора должен проходить очень быстро, за доли секунды;
· переход должен вызываться единственным фактором, одним и тем же у всех индикаторов данного типа. Так, изменение окраски кислотно-основного индикатора не должно происходить за счет реакций другого типа, например при взаимодействии с окислителями, или ионами металлов, или белками! Напротив, редокс-индикаторы должны менять свою окраскутолько вследствие взаимодействия с окислителями и восстановителями, и происходить это должно при определенном потенциале, специфическом для каждого редокс-индикатора. Окраска этих индикаторов и потенциал перехода не должны зависеть от рН раствора. К сожалению, на практике потенциал перехода многих редокс-индикаторов зависит иот рН.
Чтобы ослабить влияние побочных процессов, иногда индикатор не вводят в титруемый раствор, а, наоборот, в ходе титрования периодически отбирают каплю титруемого раствора, смешивают ее на часовом стекле с каплей раствора индикатора и наблюдают, какая окраска получается. Такой прием позволяет использовать необратимо реагирующие индикаторы. С «внешним индикатором» удобнее работать, если заранее пропитать имбумагу.
Конечная точка титрования,фиксируемая по переходу окраски индикатора, может не совпадать с точкой эквивалентности. Несовпадение V к.т.т. и V т.экв приводит к систематической погрешностирезультата анализа. Величина погрешности определяется природой данного индикатора, его концентрацией и составом титруемого раствора.
Принцип подбора индикаторов очень прост и универсален:характеристика перехода индикатора (рТ-показатель титрования, потенциал перехода и т.п.) должна соответствовать ожидаемому составу титруемого растворав точке эквивалентности. Так, если аналитик титрует водный раствор сильной кислоты сильным основанием, в точке эквивалентности раствор будет иметь рН = 7. Следовательно,надо использоватькислотно-основной индикатор, который меняет свою окраску приблизительно при рН 7 (бромтимоловый синий и т.п.).Необходимые сведения о рТ - показателях титрования для индикаторов разного типа есть в справочной литературе.
Расчет результатов титриметрического анализа
Результаты титриметрического анализа не рекомендуется рассчитывать непосредственно по уравнению реакции, например, с помощью пропорций. Такой «школьный» способ решения расчетных задач нерационален и, как правило,не дает требуемой точности. Результаты титриметрического анализа рассчитывают по одной из несколькихготовых алгебраических формул, выведенных на основании закона эквивалентов. Исходными данными будут oбъем затраченного титранта (в миллилитрах) и концентрация титранта (в моль/литр), их надо установить с необходимой точностью.
Способ расчета не зависит от типа химическойреакции, протекающейв ходе титрования, и способа контроляточки эквивалентности (индикатор, прибор и т.п.). Выбор расчетной формулы определяется тем,какойспособ титрования(прямое, обратное, заместительное) применяютв ходе анализа.Выбираяформулу, следует различать два случая:а) расчетконцентрациираствора Х;б) определениемассовой доли компонента (процентного содержанияХ в пробе).
Наиболее просто выглядят расчетные формулы, если концентрацииопределяемогокомпонента и титранта выражают числоммолей их эквивалентов в литре соответствующих растворов, т.е. используют концентрации определяемого компонента (N x ) и титранта (N T ), выраженные числом молей эквивалента в литре раствора. Ранее эти концентрации называли нормальными. Теперь этот термин применять не рекомендуется, но на практике его используют весьма широко, особенно в редоксметрии. А вот в комплексонометрии и в некоторых других методах, где 1 моль определяемого вещества Х всегда реагирует с 1 молем титранта, нормальные концентрации совпадают с обычными молярными концентрациями (C x и С Т ), а поэтому при расчете результатов нормальные концентрации и эквиваленты применять незачем.
В отличие от обычных молярных концентраций, нормальная концентрация определяется с учетом химизма реакции, протекающей в ходе титрования. Полезно запомнить, чтонормальная концентрация Х в растворе либо равнаего молярной концентрации,либо превосходит ее в несколько (2,3,4....)раз,смотря по тому, сколько протонов (или электронов) участвует в реакции, в расчете на одну частицу Х. При записи уравнения реакции, определенииэквивалентов и расчете нормальных концентраций следуетучитывать условия, в которых протекает титрование, и даже выбор индикатора.
Масса оттитрованного Xпри прямомтитровании равна (в мг):
m x =N T . V T . Э x , (1),
где Э x - молярная масса эквивалента Х, соответствующая одному протону (в кислотно-основных реакциях),одному электрону (в окислительно-восстановительных реакциях),одномулиганду (в реакциях комплексообразования), и т.п. V T – объем титранта (в мл). В комплексонометриимассу определяемого вещества (в мг) лучше рассчитывать по формуле, в которую входит величина М х -молярная масса Х:
m x = С T . V T . М x (2).
Из (4.11) следует, что массовая доля Х в навеске пробы, выраженная в %, равна:
%X = N T . V T . Э x . 100 % / m S , (3),
где m S - масса навески в мг.Обычно результат титрования не зависит от того, в какомобъеме воды растворили навеску пробы перед титрованием, и этот объем в расчетах не учитывают. Если же титруют невсю навеску, анекоторую ее часть (аликвоту), то надо учесть дополнительный коэффициент К , равныйотношению V 0 -объема раствора,в который перевелиэту навеску и из которого отбирали аликвоты,к V aliq - объемуодной аликвоты:
m x = К. N T . V T . Э x , (4).
При расчете концентрации по способу прямого (или заместительного) титрованияприменяютпростую формулу, непосредственно следующую из закона эквивалентов:
N х . V х =N T . V T (5).
анализа, однако в заводских лабораториях пользуются и другими способами расчета.
Приготовление рабочих растворов в титриметрии
Применяемые в титриметрическом анализе рабочие растворы точно известной концентрации готовят несколькими способами:
· по точной навеске химического реактива , взятой на аналитических весах. Эту навеску растворяют в небольшом количестве растворителя, а затем в мерной колбе доводят объем полученного раствора до метки. Полученныерастворыназывают стандартными, а соответствующие реактивы – первичнымистандартами. Лишь немногие вещества могут быть первичными стандартами – они должны быть чистыми химическими веществами постоянного и точно известного состава, твердыми при комнатной температуре, устойчивыми на воздухе, не гигроскопичными и не летучими. Примерами могут бытьдихромат калия, комплексон III , щавелевая кислота. Напротив, по навеске нельзя приготовить стандартный раствор соляной кислоты (реактив «соляная кислота» - жидкость с неточно известным составом), хлорида двухвалентного железа (быстро окисляется на воздухе), едкого натра (гигроскопичен) и многих других веществ.
· из фиксаналов . Этим термином называют запаянную стеклянную ампулу, в которой содержится определенное количество реагента, обычно 0,1000 моль эквивалента. Фиксаналы готовят в заводских условиях. Если в лаборатории количественно перенести содержимое фиксанала в мерную колбу на 1000 мл и довести растворителем до метки, получится литр точно 0,1000 н раствора. Приготовление фиксанальных растворов не только экономит время аналитика, но ипозволяет готовить растворы с точно известной концентрацией из таких веществ, которые не обладаюткомплексом свойств, необходимых для первичных стандартов (например, фиксанальные растворы соляной кислоты,аммиака или иода).
· по приблизительно известной навеске химического реактива, взятой на технических весах. Эту навеску растворяют в приблизительно известном количестве растворителя. Затемпроводят дополнительную операцию – стандартизацию полученного раствора. Например, титруют полученным раствором точную навеску другого вещества (первичного стандарта). Можно поступить и по-другому: взять известный объем (аликвоту) приготовленного раствора и оттитровать его подходящим стандартным раствором.По объему, пошедшему на титрование, рассчитывают точную концентрацию приготовленного раствора. Такие растворы называют стандартизованными. Например, раствор КОН стандартизуют по навеске щавелевой кислоты или с помощью фиксанального раствора соляной кислоты. Если вещество в лабораторииимеется в виде концентрированного раствора приблизительно известной концентрации (например, соляная кислота), то вместо его взвешивания отмеривают некоторый, заранее рассчитанный объем концентрированного раствора. Это требует знания плотности исходного раствора. Затем, как и в предыдущем случае, стандартизуют полученный раствор.
Концентрация растворов не должна самопроизвольно изменяться при хранении. В этом случае заранее приготовленные (стандартные или стандартизованные) растворы можно будет использовать для проведения титрованийбез каких-либо дополнительных операций.Следует учесть, что чем более разбавлен раствор, тем, как правило, он менее устойчив при хранении (гидролиз растворенного вещества, его окисление кислородом воздуха, адсорбция на внутренней поверхности стеклянной посуды и др.). Поэтомурабочие растворыс низкой концентрацией, как правило, не готовят заранее. Их готовят лишь по мере надобности, в день употребления. Для этого разбавляют исходные (стандартные, фиксанальные или стандартизованные) растворы чистым растворителем в точно известное число раз (обычно за одну операцию раствор разбавляют в 5 или 10 раз). Если требуются еще более разбавленные растворы, то эту операцию повторяют. Например, из 0,1 М раствора готовят 0,01 М, из того - 0,001 М и т.д.
Приготовление растворов с точно известной концентрацией требует использованияцелого набора специальной мерной посуды, позволяющейизмерять объемы с требуемой точностью. Это мерные колбы, пипетки и бюретки. В руководствахклабораторным работам приводятся описания мерной посудыи правила работы с ней.
Методы титрования
Методотдельных навесок и метод аликвот . Дляуменьшениявлияния случайных погрешностей титрование обычно повторяют несколько раз, а затем усредняют результаты. Повторные анализыможно проводить двумя разными способами:по методу отдельных навесоки по методу аликвот. Оба способа используют и при стандартизации рабочих растворов, и непосредственнов анализереальных объектов.
Метод отдельных навесок , как ясно из его названия, предполагает, что для титрования берут несколько навесок анализируемого материала. Массы их должны быть приблизительно равны. Размер навески выбирают с учетом желаемого расхода титранта на одно титрования (не более объема бюретки) и с учетом концентрации титранта.
Пусть взяты три навески щавелевой кислоты,массы которыхуказаны в табл.2. По данным каждого титрования вычисляют (по отдельности!) концентрацию КОН. Затем усредняютконцентрации.Объемы, затраченные на титрование разных навесок,усреднять нельзя!
Таблица 2.Пример расчета результатов анализа по методу отдельных навесок
|
Номернавески |
Массанавески,мг |
Объемтитранта,мл |
Найденная концентрацияКОН, моль/л |
|
95,7 |
14,9 |
0,102 |
|
|
106,9 |
16,2 |
0,105 |
|
|
80,8 |
12,7 |
0,101 |
|
|
Средний результат анализаС КОН =0,103 моль/л |
|||
Метод титровпанияаликвот (или метод пипетирования) основан на титровании нсекольких отдельных аликвот – небольших объемов исследуемого раствора, отобранных с помощью пипеток.
Метод отдельных навесок и методтитрованияаликвотиспользуют не только при прямом титровании, как это показано в приведенных примерах, но и при обратном, и при заместительном титровании. Выбирая способ титрования, следует учесть, что метод отдельных навесок дает более точные результаты, но он более трудоемкий и требуетбольшего объема расчетов. Поэтому метод отдельных навесок лучше использоватьдля стандартизации рабочих растворов, а для серийно выполняемыханализов применять более экспрессный метод аликвот.
Форма кривых титрования
Логарифмические кривые титрования представляют графическую зависимостьлогарифмаравновеснойконцентрации одного из реагентов от объема добавленного титранта. Вместо логарифма концентрации на вертикальной оси обычно откладывают величину рН раствора (водородный показатель). Применяют и другие аналогичные показатели (например, pAg = - lg ), а также величину тех физико-химических свойств титруемого раствора, которые линейно зависят от логарифмов равновесных концентраций. Примером может быть электродный потенциал (E ).
Если в растворе содержитсятолько одно вещество, реагирующее с титрантом, причемреакция описывается единственным химическим уравнением (то есть проходит не ступенчато)- на логарифмической кривой наблюдается почти вертикальный участок, называемой скачком титрования . Напротив, участкикривой вдали от т.экв. близки к горизонтальным. Примером могут быть зависимости рН растворов от объема V добавленного титранта, показанные на рис.1
Рис.1. Вид кривых титрования
Чем выше высота скачка на кривой тирования, тем точнее можно зафиксировать точку эквивалентности.
Кислотно-основное титрование (метод нейтрализации)
Принцип метода
Метод нейтрализации основан на проведении кислотно-основных (протолитических) реакций. В ходе такого титрования меняетсязначение рН раствора. Кислотно-основные реакции подходят для титриметрического анализа в наибольшей степени: они протекают по строго определенным уравнениям, без побочных процессов и с очень высокой скоростью. Взаимодействие сильных кислот с сильными основаниями приводит к высоким константам равновесия. Для обнаружения к.т.т. существует удобный и хорошо изученный способ - применение кислотно-основных индикаторов. Можно использовать и инструментальные методы, они особенно важны при титровании неводных, мутныхили окрашенных растворов.
Метод нейтрализации включаетдва варианта – ацидиметрию (титрант – раствор сильной кислоты) и алкалиметрию (титрант – раствор сильного основания). Эти методы соответственно применяют для определения оснований и кислот, в том числе ионных и многопротонных. Возможность титрования сильных протолитов определяется их концентрацией; титрование возможно, если С х > 10 - 4 М .В ходе такого титрованияв водном растворе идет реакция:
H 3 O + +OH - ® 2 Н 2 О
Титрование слабых кислот и слабых оснований в водных растворах соответствует схемам:
НА+ОН - ® Н 2 О(алкалиметрия)
В+Н 3 O + ® НВ + + Н 2 О(ацидиметрия)
Примеры практического применения кислотно-основного титрования:
· определение кислотности пищевых продуктов, почв и природных вод (алкалиметрическое титрованиеводных растворов с индикатором фенолфталеином);
· определение кислотности нефтепродуктов (алкалиметрическое титрование неводных растворов с инструментальным контролем к.т.т.);
· определение карбонатов и гидрокарбонатов в минералах и строительных материалах (ацидиметрическое титрование водных растворов с двумя индикаторами);
· определение азота в солях аммония и в органических веществах (метод Кьельдаля). В этом случае органические азотсодержащие вещества разлагают кипячением с концентрированной серной кислотой в присутствии солей ртути, аммонийный азот отгоняют действием щелочи при нагревании, аммиак поглощают стандартным раствором НСl , взятым в избытке. Затем титруют щелочью непрореагировавшую часть НСl в присутствии индикатора метилового оранжевого. В данной методике используют и принцип замещения, и способ обратного титрования.
Рабочие растворы. При ацидиметрическом титровании водных растворовв качестве титрантов используют растворы сильных кислот (НСl , реже НNO 3 или H 2 SO 4). В алкалиметрии титранты - растворы NaOH или КОН. Однако перечисленные реагенты не обладают свойствами, которые позволяли бы готовитьиз них стандартные растворыпросто по точной навеске. Так, твердые щелочи гигроскопичны и всегда содержат примеси карбонатов. В случае НСl и других сильных кислот исходный реактив представляет собой не чистое вещество, а раствор с неточно известной концентрацией. Поэтому в методе нейтрализации вначале готовят раствор с приблизительно известной концентрацией, а потомстандартизуют его. Растворы кислот стандартизуют по безводному карбонату натрия Na 2 CO 3 (соде) или по тетраборату натрия Na 2 B 4 O 7 . 10Н 2 О (буре). Бура при растворении взаимодействует с водой:
В 4 О 7 2– +3Н 2 О=2Н 3 ВО 3 + 2ВО 2 –
Образовавшийся метаборат - довольно сильное основание. Его титруют кислотой:
ВО 2 – + Н 3 О + = Н 3 ВО 3 .
Очевидно, что молярная масса эквивалента буры равна М (½Na 2 B 4 O 7 . 10Н 2 О) = 190.71 г/моль. Высокая молярная масса эквивалента – преимущество буры как первичного стандарта. Растворы щелочей стандартизуют по гидрофталату калия. Молекулагидрофталатасодержит подвижный протон и обладает свойствами слабой кислоты:
В качестве стандартов нередко используют бензойную кислоту С 6 Н 5 СООН, щавелевую кислоту H 2 C 2 O 4 . 2H 2 O и другие слабые органические кислоты (твердые, чистые устойчивые вещества). Стандартные 0,1000 М растворы кислот и оснований в лабораториях обычно готовят из фиксаналов. Приготовленный раствор кислоты можно использовать для стандартизации раствора щелочи, и наоборот. Стандартизованные растворы кислот устойчивы и могут храниться без изменения сколь угодно долго. Растворы щелочей менее устойчивы, рекомендуется хранить их в парафинированной или фторопластовой посуде, чтобы не допустить взаимодействия со стеклом. Необходимо учитывать, что растворы щелочей поглощают СО 2 из воздуха, при хранении их защищают с помощью трубки, заполненной негашеной или натронной известью.
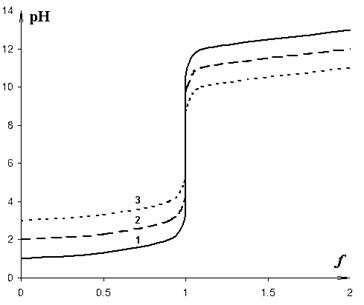
Рис. 2. Кривые нейтрализации сильной кислоты.
1 - 0,1 М, 2 - 0,01 М, 3 – 0,001 М.
Для обнаружения к.т.т. с цветным индикатором необходимо, чтобы высота скачкабыла больше, чем ширина интервала перехода индикатора. Последняя обычно составляет около двух единиц рН.
Высота скачка на кривой нейтрализации слабых кислот зависит отсилы кислоты(величины ее кислотной константы, или рK a ). А именно, чем слабее кислота (чем больше величина рК а), тем меньше при прочих равных условиях должны быть высота скачка.разной силы
1 -соляная кислота,2 – уксусная кислота (рК а = 4,8),3 – синильная кислота (pK a = 9,2).
Высота скачка должна быть большеширины зоны перехода индикатора, которая, как правило, составляет 2 единицы рН. Поэтому,как и в случае сильных электролитов, критерий возможностититрования слабого протолита с 1 %-ной ошибкой можно вывести из условия ∆p Н ±1% ≥ 2. Для водного раствора слабой кислоты получаем искомый критерий в следующей форме:
рК a + рС ≤ 8
Приp С = 2критическое значение рК а равно 6. Иными словами, если кислота очень слабая, и ее рК а больше 6, то точно оттитровать ее с цветными индикаторами нельзя.
Титрование смесей протолитов и многопротонных протолитов. В смешанных растворахсильные кислоты подавляют протолиз более слабых. То же наблюдается в растворах, содержащих смесь оснований разной силы.При добавлении к такой смеси титранта прежде всего оттитровывается более сильный протолит, а уже затем с титрантом реагирует более слабый. Однако число скачков, наблюдаемых на кривой титрования смеси, зависитне только от числа присутствующих протолитов, но и от абсолютных значений соответствующих констант кислотности (основности), а также от их соотношения. Константы кислотности (или основности) компонентов смеси должны различаться более чем в 10 4 ,раз, только в этом случае на кривой титрования будутраздельно наблюдатьсяотчетливо выраженные скачки титрования, а относительная ошибка определения каждого компонентане превысит 1 %. Критерием возможности раздельного титрования протолитов является так называемое «правило четырех единиц»:
![]() (6)
(6)
Многопротонные протолиты реагируют с титрантамиступенчато, сначала по первой ступени, затем по второй и т.д., если соответствующие константы кислотности различаются в соответствии с условием (6).При расчете кривых нейтрализации многопротонные протолиты можно рассматривать каксмесиразных электролитов.
В качестве примера проанализируем возможность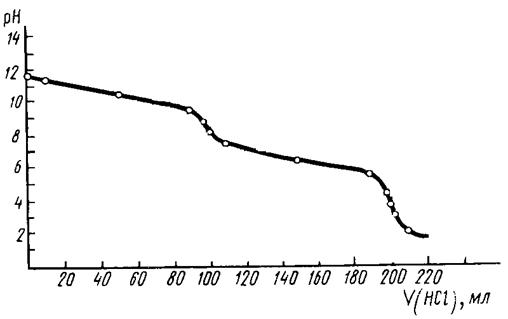
Рис.5. Кривая титрования смеси карбонат- и гидрокарбонат-ионов раствором HCl .
Указаны значения рН, при которых наблюдаются переходы окраски индикаторов.
При титровании смеси двух сильных кислот, смеси двух одинаково слабых кислот или смеси двух оснований с близкими рК b двух раздельных скачков на кривой титрования нет. Однако определить концентрацию компонентов таких смесей по отдельности все же вполне возможно. Эти задачи успешно решают, используя дифференцирующие неводные растворители.
Кислотно-основные индикаторы и их выбор
Для обнаружения к.т.т. в методе нейтрализации традиционно используют кислотно-основные индикаторы – синтетические органические красители, являющиеся слабыми кислотами или основаниями и меняющие видимую окраску в зависимости от рН раствора.Примеры некоторых (наиболее часто применяемых в лабораториях) кислотно-основных индикаторов приведены в таблице 3. Строение и свойства индикаторов приведены в справочниках. Важнейшими характеристиками каждого кислотно-основного индикатора являются интервал перехода и показатель титрования (pT ). Интервал перехода – это зона между двумя значениями рН, соответствующими границам зоны, внутри которой наблюдается смешанная окраска индикатора. Так водный раствор метилового оранжевого наблюдатель охарактеризует как чисто желтый – при рН< 3,1 и как чисто красный при рН > 4,4, а между этими граничными значениями наблюдается смешанная, розово-оранжевая окраска разных оттенков. Ширина интервала перехода обычно составляет 2 единицы рН. Экспериментально определенные интервалы перехода индикаторов в некоторых случаях меньше или больше двух единиц рН. Это, в частности, объясняется различной чувствительностью глаза к разным участкам видимой области спектра. Для одноцветных индикаторов ширина интервала зависит и от концентрациииндикатора.
Таблица 3
Важнейшие кислотно-основные индикаторы
|
Индикатор |
Интервалперехода ΔрН Ind |
рК a (HInd ) |
Изменение окраски |
|
|
Метиловый оранжевый |
Красная - желтая |
|||
|
Бромкрезоловый зеленый |
Желтая - синяя |
|||
|
Метиловый красный |
Красная - желтая |
|||
|
Бромкрезоловый пурпурный |
Желтая - фиолетовая |
|||
|
Бромтимоловый синий |
Желтая - синяя |
|||
|
Феноловый красный |
Желтая - красная |
|||
|
Тимоловый синий |
||||
|
Фенолфталеин |
Бесцветная - красная |
Зная характеристики разных индикаторов, можно теоретически обоснованно подбирать их,чтобы получить правильные результаты анализа.Придерживаются следующего правила: интервал перехода индикатора должен лежать в области скачка на кривой титрования .
При выборе индикаторов для титрования слабых протолитов необходимо учитывать, что т.экв. и скачок титрования смещены в слабощелочную среду при титровании кислоты и в слабокислую среду – при титровании основания. Следовательно, для титрования слабых кислот подходят индикаторы, меняющие свою окраску в слабощелочной среде (например, фенолфталеин), а для титрования слабого основания – индикаторы, меняющие окраску в слабокислой среде (например, метиловый оранжевый
Существует ещё одна характеристика каждого кислотно-основного индикатора –это показатель титрования (рТ). Так называют значение рН, при котором наблюдатель наиболее отчетливо замечает изменение окраски индикатора и именно в этот момент считает титрование законченным. Очевидно, рТ = рН К.Т.Т. .Выбирая подходящий индикатор, надо стремиться к тому, чтобы величина рТ была бы как можно ближе ктеоретически рассчитанной величине рН Т.ЭКВ.. Обычно значение рТблизко к середине интервала перехода. Но рT – плохо воспроизводимая величина. Разные люди, проводящие одно и то же титрование с одним и тем же индикатором, получат существенно различные значения pT .К тому же величина рТ зависит от порядка титрования, то есть от направления изменения окраски.При титровании кислот и оснований с одним и тем же индикатором значения рТбудут несколько различаться. Для одноцветных индикаторов (фенолфталеин и т.п.) величина рТ зависит и от концентрации индикатора.
Введение
Лабораторный практикум выполняется после изучения теоретического курса «Аналитическая химия и ФХМА» и служит для закрепления и углубления полученных знаний.
Задачей количественного анализа является определение количества (содержания) элементов (ионов), радикалов, функциональных групп, соединений или фаз в анализируемом объекте . В этом курсе рассматриваются основные методы титриметрического (объемного) анализа, способы титрования и их практическое применение.
Прежде чем приступить к выполнению лабораторного практикума, студенты проходят инструктаж по технике безопасности. Перед выполнением каждой работы студент должен сдать коллоквиум по разделам, указанным преподавателем, а также по методике проведения анализа. Для этого необходимо:
1) повторить соответствующий раздел курса;
2) подробно ознакомиться с методикой проведения работы;
3) составить уравнения химических реакций, лежащих в основе проводимого химического анализа;
4) изучить особенности проведения анализа с точки зрения техники безопасности.
По результатам работы студенты составляют отчёт, в котором должны быть указаны:
· название работы;
· цель работы;
· теоретические основы метода: сущность метода, основное уравнение, расчеты и построение кривых титрования, выбор индикатора;
· реактивы и оборудование, используемые в ходе проведения работы;
· методика анализа:
Приготовление первичных стандартов;
Приготовление и стандартизация рабочего раствора;
Определение содержания исследуемого вещества в растворе;
· экспериментальные данные;
· статистическая обработка результатов анализа;
· выводы.
ТИТРИМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Титриметрический метод анализа основан на измерении объема реагента точно известной концентрации (титранта), затраченного на химическую реакцию с определяемым веществом.
Процедура определения (титрование) состоит в том, что к точно известному объему раствора определяемого вещества с неизвестной концентрацией из бюретки по каплям добавляют титрант, до наступления точки эквивалентности.
где X – определяемое вещество; R – титрант, P – продукт реакции.
Точка эквивалентности (т.э.) – это теоретическое состояние раствора, наступающее в момент добавления эквивалентного количества титранта R к определяемому веществу X . На практике титрант добавляют к определяемому веществу до достижения конечной точкой титрования (к.т.т.), под которой понимают при визуальной индикации точки эквивалентности момент изменения окраски индикатора, добавленного в раствор. Кроме визуальной индикации точка эквивалентности может быть зарегистрирована инструментальными способами. В этом случае под конечной точкой титрования (к.т.т.) понимают момент резкого изменения физической величины, измеряемой в процессе титрования (сила тока, потенциал, электропроводность и т. д.).
В титриметрическом методе анализа используются следующие типы химических реакций: реакции нейтрализации, реакции окисления-восстановления, реакции осаждения и реакции комплексообразования.
В зависимости от типа применяемой химической реакции различают следующие методы титриметрического анализа:
– кислотно-основное титрование;
– осадительное титрование;
– комплексонометрическое титрование или комплексонометрия;
– окислительно-восстановительное титрование или редоксиметрия.
К реакциям, применяемым в титриметрическом методе анализа, предъявляют следующие требования:
· реакция должна протекать в стехиометрических соотношениях, без побочных реакций;
· реакция должна протекать практически необратимо (≥ 99,9 %), константа равновесия реакции К р >10 6 , образующиеся осадки должны иметь растворимость S < 10 -5 моль/дм 3 , а образующиеся комплексы – К уст > 10 -6 ;
· реакция должна протекать с достаточно большой скоростью;
· реакция должна протекать при комнатной температуре;
· точка эквивалентности должна фиксироваться четко и надежно каким-либо способом.
Способы титрования
В любом методе титриметрического анализа существует несколько способов титрования. Различают прямое титрование, обратное титрование и титрование по замещению .
Прямое титрование – к раствору определяемого вещества добавляют по каплям титрант до достижения точки эквивалентности.
Схема титрования: X + R = P .
Закон эквивалентов для прямого титрования:
C (1/ z) Х V Х = C (1/ z) R V R . (2)
Количество (массу) определяемого вещества, содержащееся в исследуемом растворе, вычисляют, используя закон эквивалентов (для прямого титрования)
m Х = C (1/z)R V R M (1/z) Х ٠10 -3 , (3)
где C (1/ z) R – молярная концентрация эквивалента титранта, моль/дм 3 ;
V R – объем титранта, см 3 ;
M (1/ z ) Х – молярная масса эквивалента определяемого вещества;
C (1/ z) Х – молярная концентрация эквивалента определяемого вещества, моль/дм 3 ;
V Х – объем определяемого вещества, см 3 .
Обратное титрование
– используют два титранта. Сначала
к анализируемому раствору добавляют точный объем первого титранта (R 1
), взятый в избытке. Остаток непрореагировавшего титранта R 1 оттитровывают вторым титрантом (R 2
). Количество титранта R 1
, израсходованного
на взаимодействие с анализируемым веществом (Х
) определяют по разности между добавленным объемом титранта R 1
(V 1
) и объемом титранта R 2
(V 2
) затраченного на титрование остатка титранта R 1
.
Схема титрования: X + R 1 фиксированный избыток = P 1 (R 1 остаток).
R 1 остаток + R 2 = P 2 .
При использовании обратного титрования закон эквивалентов записывается следующим образом:
Массу определяемого вещества в случае обратного титрования вычисляют по формуле
Способ обратного титрования применяется в тех случаях, когда для прямой реакции невозможно подобрать подходящий индикатор или она протекает с кинетическими затруднениями (низкая скорость химической реакции).
Титрование по замещению (косвенное титрование) – применяют в тех случаях, когда прямое или обратное титрование определяемого вещества невозможно или вызывает затруднения либо отсутствует подходящий индикатор.
К определяемому веществу Х добавляют какой-либо реагент А в избытке, при взаимодействии с которым выделяется эквивалентное количество вещества Р . Затем продукт реакции Р оттитровывают подходящим титрантом R .
Схема титрования: X + А избыток = P 1.
P 1 + R = P 2.
Закон эквивалентов для титрования по замещению записывают следующим образом:
Так как число эквивалентов определяемого вещества Х и продукта реакции Р одинаковы, расчет массы определяемого вещества в случае косвенного титрования вычисляют по формуле
m Х = C (1/z) R V R M (1/z) Х ٠10 -3 . (7)
Реактивы
1. Янтарная кислота Н 2 С 4 Н 4 О 4 (х.ч.) – первичный стандарт.
2. Раствор гидроксида натрия NaOH с молярной концентрацией
~2,5 моль/дм 3
3. Н 2 О дистиллированная.
Оборудование студенты описывают самостоятельно.
Ход выполнения работы:
1. Приготовление первичного стандарта янтарной кислоты HOOCCH 2 CH 2 COOH.
Янтарную кислоту готовят объемом 200,00 см 3 с молярной концентрацией эквивалента ![]() моль/дм 3 .
моль/дм 3 .
![]() г/моль.
г/моль.
Уравнение реакции:
Взятие навески (взвешивание):
![]()
Масса навески
Навеску количественно
переносят в мерную колбу (![]() см 3), добавляют 50 – 70 см 3 дистиллированной воды, перемешивают до полного растворения янтарной кислоты, доводят до метки дистиллированной водой
см 3), добавляют 50 – 70 см 3 дистиллированной воды, перемешивают до полного растворения янтарной кислоты, доводят до метки дистиллированной водой
и тщательно перемешивают.
рассчитывают
по формуле

Реактивы
1. Карбонат натрия Na 2 CO 3 (х.ч.) – первичный стандарт.
2. Н 2 О дистиллированная.
3. Хлороводородная кислота НСl концентрации 1:1 (r=1,095 г/см 3).
4. Кислотно-основной индикатор (выбирают по кривой титрования).
5. Смешанный индикатор – метиловый оранжевый и метиленовый синий.
Ход выполнения работы:
1. Приготовление первичного стандарта карбоната натрия (Na 2 CO 3).
Раствор карбоната натрия готовят объёмом 200,00 см 3 с молярной концентрацией эквивалента ![]() моль/дм 3 .
моль/дм 3 .
Расчет массы навески, г: (масса берется с точностью до четвертого знака после запятой).
Уравнения реакции:
1) Na 2 CO 3 + HCl = NaHCO 3 + NaCl
2) NaHCO 3 + HCl = NaCl + H 2 O + CO 2
_____________________________________
Na 2 CO 3 +2HCl = 2NaCl + H 2 O + CO 2
H 2 CO 3 – слабая кислота (K a1 = 10 -6,35 , K a2 = 10 -10,32).
Взятие навески (взвешивание):
Масса часового стекла (стакана)
Масса часового стекла (стакана) с навеской ![]()
Масса навески
Навеску количественно
переносят в мерную колбу (![]() см 3), добавляют 50 – 70 см 3 дистиллированной воды, перемешивают до полного растворения карбоната натрия, доводят до метки дистиллированной водой
см 3), добавляют 50 – 70 см 3 дистиллированной воды, перемешивают до полного растворения карбоната натрия, доводят до метки дистиллированной водой
и тщательно перемешивают.
Фактическую концентрацию первичного стандарта
рассчитывают
по формуле

2. Приготовление и стандартизация титранта (раствора HCl)
Раствор хлороводородной кислоты готовят объемом примерно 500 см 3
с молярной концентрацией эквивалента примерно 0,05÷0,06 моль/дм 3)
Титрант – раствор хлороводородной кислоты приблизительной концентрацией 0,05 моль/дм 3 готовят из хлороводородной кислоты, разбавленной 1:1 (r=1,095 г/см 3).
Стандартизацию раствора HCl проводят по первичному стандарту Na 2 CO 3 прямым титрованием, способом пипетирования.
Индикатор выбирают по кривой титрования карбоната натрия хлороводородной кислотой (рис. 4).

Рис. 4. Кривая титрования 100,00 см 3 раствора Na 2 CO 3 с С = 0,1000 моль/дм 3 раствором HCl с С 1/ z = 0,1000 моль/дм 3
При титровании до второй точки эквивалентности используют индикатор метиловый оранжевый, 0,1%-ный водный раствор (рТ = 4,0). Изменение окраски от желтой до оранжевой (цвет «чайной розы»). Интервал перехода
(рН = 3,1 – 4,4) .
Схема 3. Стандартизация раствора HCl

В коническую колбу для титрования вместимостью 250 см 3 помещают аликвоту 25,00 см 3 стандартного раствора Na 2 CO 3 (пипеткой), добавляют 2 – 3 капли метилового оранжевого, разбавляют водой до 50 – 75 см 3 и титруют раствором хлороводородной кислоты до перехода окраски из желтой в цвет «чайной розы» от одной капли титранта. Титрование проводят в присутствии «свидетеля» (исходный раствор Na 2 CO 3 с индикатором). Результаты титрования заносят в табл. 4. Концентрацию хлороводородной кислоты определяют по закону эквивалентов: .
Таблица 4
Результаты стандартизации раствора соляной кислоты
Задачи
1. Сформулируйте понятие эквивалента в кислотно-основных реакциях . Вычислите величину эквивалентов соды и фосфорной кислоты в следующих реакциях:
Na 2 CO 3 + HCl = NaHCO 3 +NaCl
Na 2 CO 3 + 2HCl = 2NaCl + CO 2 + H 2 O
H 3 PO 4 + NaOH = NaH 2 PO 4 + H 2 O
H 3 PO 4 + 2NaOH = Na 2 HPO 4 + H 2 O
H 3 PO 4 + 3NaOH = Na 3 PO 4 + 3H 2 O
2. Напишите уравнения реакций между соляной кислотой, серной кислотой, гидроксидом натрия, гидроксидом алюминия, карбонатом натрия, гидрокарбонатом калия и рассчитайте эквивалентную массу этих веществ.
3. Постройте кривую титрования 100,00 см 3 соляной кислоты с молярной концентрацией эквивалента 0,1 моль/дм 3 гидроксидом натрия с молярной концентрацией эквивалента 0,1 моль/дм 3 . Выберите возможные индикаторы
4. Постройте кривую титрования 100,00 см 3 акриловой кислоты (CH 2 =CHCOOH, pK a
= 4,26) с молярной концентрацией эквивалента
0,1 моль/дм 3 гидроксидом натрия с молярной концентрацией эквивалента
0,1 моль/дм 3 . Как изменяется состав раствора в процессе титрования? Выберите возможные индикаторы и рассчитайте индикаторную погрешность титрования.
5. Постройте кривую титрования гидразина (N 2 H 4 +H 2 O, pK b
= 6,03)
с молярной концентрацией эквивалента 0,1 моль/дм 3 соляной кислотой
с молярной концентрацией эквивалента 0,1 моль/дм 3 . В чем сходство
и различие расчетов рН и кривой титрования в сравнении с кривой титрования слабой кислоты щелочью? Выберите возможные индикаторы
и рассчитайте индикаторную погрешность титрования.
6. Вычислите коэффициенты активности и активные концентрации ионов
в 0,001 М растворе сульфата алюминия, 0,05 М карбоната натрия, 0,1 М хлорида калия.
7. Вычислите рН 0,20 М раствора метиламина, если его ионизация в водном растворе описывается уравнением
В + Н 2 О = ВН + + ОН - , К b = 4,6 ×10 - 3 , где В – основание.
8. Вычислить константу диссоциации хлорноватистой кислоты HOCl, если 1,99 × 10 - 2 М раствор имеет рН = 4,5.
9. Вычислите рН раствора, содержащего 6,1 г/моль гликолевой кислоты (СH 2 (OH)COOH, К а = 1,5 × 10 - 4).
10. Вычислите рН раствора, полученного смешением 40 мл 0,015 М раствора хлороводородной кислоты с:
а) 40 мл воды;
б) 20 мл 0,02 М раствора гидроксида натрия;
в) 20 мл 0,02 М раствора гидроксида бария;
г) 40 мл 0,01 М раствора хлорноватистой кислоты, К а =5,0 × 10 - 8 .
11. Вычислите концентрацию ацетат-иона в растворе уксусной кислоты
c массовой долей 0,1 %.
12. Вычислите концентрацию иона аммония в растворе аммиака c массовой долей 0,1 %.
13. Рассчитайте массу навески карбоната натрия, необходимую для приготовления 250,00 мл 0,5000 М раствора .
14. Рассчитайте объем раствора соляной кислоты с молярной концентрацией эквивалента 11 моль/л и объем воды, которые необходимо взять для приготовления 500 мл 0,5 М раствора соляной кислоты.
15. В 300 мл 0,3 %-ного раствора хлороводородной кислоты растворили 0,15 г металлического магния. Вычислите молярную концентрацию ионов водорода, магния и хлора в полученном растворе.
16. При смешении 25,00 мл раствора серной кислоты с раствором хлорида бария получено 0,2917 г сернокислого бария. Определите титр раствора серной кислоты.
17. Вычислить массу карбоната кальция, вступившего в реакцию
с 80,5 ммоль хлороводородной кислоты.
18. Сколько граммов однозамещенного фосфата натрия надо добавить
к 25,0 мл 0,15 М раствора гидроксида натрия, чтобы получить раствор с рН=7? Для фосфорной кислоты pK а1
= 2,15; pK а2
= 7,21; pK а3 =
12,36.
19. На титрование 1,0000 г дымящейся серной кислоты, тщательно разбавленной водой, расходуется 43,70 мл 0,4982 М раствора гидроксида натрия. Известно, что дымящаяся серная кислота содержит серный ангидрид, растворенный в безводной серной кислоте. Вычислить массовую долю серного ангидрида в дымящей серной кислоте.
20. Абсолютная погрешность измерения объема с помощью бюретки составляет 0,05 мл. Рассчитать относительную погрешность измерения объемов в 1; 10 и 20 мл.
21. В мерной колбе вместимостью 500,00 мл приготовлен раствор
из навески 2,5000 г карбоната натрия. Вычислить:
а) молярную концентрацию раствора;
б) молярную концентрацию эквивалента (½ Na 2 CO 3);
в) титр раствора;
г) титр по соляной кислоте.
22. Какой объем 10 %-ного раствора карбоната натрия плотностью
1,105 г/см 3 нужно взять для приготовления:
а) 1 л раствора с титром ТNa 2 CO 3 = 0,005000 г/см 3 ;
б) 1 л раствора с ТNa 2 CO 3 /HCl = 0,003000 г/см 3 ?
23. Какой объем соляной кислоты с массовой долей 38,32 % и плотностью 1,19 г/см 3 следует взять для приготовления 1500 мл 0,2 М раствора?
24. Какой объем воды нужно добавить к 1,2 л 0,25 М HCl, чтобы приготовить 0,2 М раствор?
25. Из 100 г технического гидроксида натрия, содержащего 3 % карбоната натрия и 7 % индифферентных примесей, приготовили 1л раствора. Вычислить молярную концентрацию и титр по соляной кислоте полученного щелочного раствора, считая, что карбонат натрия титруется до угольной кислоты.
26. Имеется образец, в котором может содержаться NaOH, Na 2 CO 3 , NaHCO 3 или смесь названных соединений массой 0,2800 г. Пробу растворили в воде.
На титрование полученного раствора в присутствии фенолфталеина расходуется 5,15 мл, а в присутствии метилового оранжевого – 21,45 мл соляной кислоты с молярной концентрацией эквивалента 0,1520 моль/л. Определить состав образца и массовые доли компонентов в образце.
27. Постройте кривую титрования 100,00 см 3 0,1000 М раствора аммиака 0,1000 М раствором соляной кислоты, обоснуйте выбор индикатора.
28. Вычислите рН точки эквивалентности, начала и конца титрования 100,00 см 3 0,1000 М раствора малоновой кислоты (HOOCCH 2 COOH) 0,1000 М раствором гидроксида натрия (рК а 1 =1,38; рК а 2 =5,68).
29. На титрование 25,00 см 3 раствора карбоната натрия с молярной концентрацией эквивалента 0,05123 моль/дм 3 пошло 32,10 см 3 соляной кислоты. Вычислите молярную концентрацию эквивалента соляной кислоты.
30. Сколько мл 0,1 М раствора хлорида аммония необходимо добавить
к 50,00 мл 0,1 М раствора аммиака, чтобы получился буферный раствор
с рН=9,3.
31. Смесь серной и фосфорной кислот перенесли в мерную колбу объемом 250,00 см 3 . Для титрования взяли две пробы по 20,00 см 3 , одну оттитровали раствором гидроксида натрия с молярной концентрацией эквивалента
0,09940 моль/дм 3 с индикатором метилоранжем, а вторую с фенолфталеином. Расход гидроксида натрия в первом случае составил 20,50 см 3 , а во втором 36,85 см 3 . Определите массы серной и фосфорной кислот в смеси.
В комплексонометрии
До точки эквивалентности =(C M V M – C ЭДТА V ЭДТА)/(V М +V ЭДТА). (21)
В точке эквивалентности = ![]() . (22)
. (22)
После точки эквивалентности =  . (23)
. (23)
На рис. 9 показаны кривые титрования иона кальция в буферных растворах с различными значениями рН. Видно, что титрование Са 2+ возможно только при рН ³ 8.
Реактивы
2. Н 2 О дистиллированная.
3. Стандартный раствор Mg (II) с молярной концентрацией
0,0250 моль/дм 3 .
4. Аммиачный буфер с рН = 9,5.
5. Раствор гидроксида калия КОН с массовой долей 5%.
6. Эриохром черный Т, индикаторная смесь.
7. Калькон, индикаторная смесь.
Теоретические основы метода:
Метод основан на взаимодействии ионов Са 2+ и Мg 2+ с динатриевой солью этилендиаминтетрауксусной кислоты (Na 2 H 2 Y 2 или Na-ЭДТА) с образованием прочных комплексов в молярном отношении M:L=1:1 в определённом интервале рН.
Для фиксирования точки эквивалентности при определении Са 2+ и Мg 2+ используют калькон и эриохром черный Т.
Определение Са 2+ проводят при рН ≈ 12, при этом Mg 2+ находится
в растворе в виде осадка гидроксида магния и не титруется ЭДТА.
Mg 2+ + 2OH - = Mg(OH) 2 ↓
Са 2+ + Y 4- « CaY 2-
При рН ≈ 10 (аммиачный буферный раствор) Мg 2+ и Са 2+ находятся
в растворе в виде ионов и при добавлении ЭДТА титруются совместно.
Ca 2+ + HY 3- « CaY 2- + H +
Mg 2+ + HY 3- « MgY 2- +H +
Для определения объема ЭДТА, затраченного на титрование Mg 2+ ,
из суммарного объёма, пошедшего на титрование смеси при рН ≈ 10, вычитают объём, пошедший на титрование Са 2+ при рН ≈ 12.
Для создания рН ≈ 12 применяют 5% – ный раствор KOH, для создания
рН ≈ 10 используют аммиачный буферный раствор (NH 3 ×H 2 O + NH 4 Cl).
Ход выполнения работы:
1. Стандартизация титранта – раствора ЭДТА (Na 2 H 2 Y)
Раствор ЭДТА готовят приблизительной концентрации 0,025 М
из ≈ 0,05 М раствора, разбавляя его дистиллированной водой в 2 раза. Для стандартизации ЭДТА применяют стандартный раствор MgSO 4
c концентрацией 0,02500 моль/дм 3 .
Схема 5. Стандартизация титранта – раствора ЭДТА
 В коническую колбу для титрования вместимостью 250 см 3 помещают 20,00 cм 3 стандартного раствора MgSO 4 c концентрацией 0,02500 моль/дм 3 , добавляют ~ 70 см 3 дистиллированной воды, ~ 10 см 3 аммиачного буферного раствора с рН ~ 9,5 – 10 и вносят индикатор эриохром чёрный Т около 0,05 г
В коническую колбу для титрования вместимостью 250 см 3 помещают 20,00 cм 3 стандартного раствора MgSO 4 c концентрацией 0,02500 моль/дм 3 , добавляют ~ 70 см 3 дистиллированной воды, ~ 10 см 3 аммиачного буферного раствора с рН ~ 9,5 – 10 и вносят индикатор эриохром чёрный Т около 0,05 г
(на кончике шпателя). При этом раствор окрашивается в винно-красный цвет. Раствор в колбе медленно титрируют раствором ЭДТА до перехода окраски из винно-красной в зелёную. Результаты титрования заносят в табл. 6. Концентрацию ЭДТА определяют по закону эквивалентов: ![]() .
.
Таблица 6
Результаты стандартизации раствора ЭДТА
2. Определение содержания Са 2+
Кривые титрования Са 2+ раствором ЭДТА при рН=10 и рН=12 строят самостоятельно.
Раствор задачи в мерной колбе доводят до метки дистиллированной водой и тщательно перемешивают.
Схема 6. Определение содержания Са 2+ в растворе
 В коническую колбу для титрования вместимостью 250 см 3 помещают аликвоту исследуемого раствора 25,00 см 3 , содержащую кальций и магний, добавляют ~ 60 см 3 воды, ~ 10 см 3 5% – ного раствора КОН. После выпадения аморфного осадка Mg(OH) 2 ↓ в раствор вносят индикатор калькон около 0,05 г (на кончике шпателя) и медленно титруют раствором ЭДТА до перехода окраски из розовой в бледно-голубую. Результаты титрования (V
1) заносят в табл.7.
В коническую колбу для титрования вместимостью 250 см 3 помещают аликвоту исследуемого раствора 25,00 см 3 , содержащую кальций и магний, добавляют ~ 60 см 3 воды, ~ 10 см 3 5% – ного раствора КОН. После выпадения аморфного осадка Mg(OH) 2 ↓ в раствор вносят индикатор калькон около 0,05 г (на кончике шпателя) и медленно титруют раствором ЭДТА до перехода окраски из розовой в бледно-голубую. Результаты титрования (V
1) заносят в табл.7.
Таблица 7
| № опыта | Объем ЭДТА, см 3 | Содержание Са 2+ в растворе, г | |
| 25,00 | |
||
| 25,00 | |||
| 25,00 | |||
| 25,00 | |||
| 25,00 |
3. Определение содержания Mg 2+
Кривую титрования Mg 2+ раствором ЭДТА при рН=10 строят самостоятельно.
Схема 7. Определение содержания Mg 2+ в растворе

В коническую колбу для титрования вместимостью 250 см 3 помещают аликвоту 25,00 см 3 исследуемого раствора, содержащую кальций и магний, добавляют ~ 60 см 3 дистиллированной воды, ~ 10 см 3 аммиачного буферного раствора с рН ~ 9,5–10 и вносят индикатор эриохром чёрный Т около 0,05 г
(на кончике шпателя). При этом раствор окрашивается в винно-красный цвет. Раствор в колбе медленно титрируют раствором ЭДТА до перехода окраски из винно-красной в зелёную. Результаты титрования (V
2) заносят в табл. 8.
Таблица 8
Результаты титрования раствора, содержащего кальций и магний
| № опыта | Объем исследуемого раствора, см 3 | Объем ЭДТА, V ∑ , см 3 | Содержание Mg 2+ в растворе, г |
| 25,00 | |||
| 25,00 | |||
| 25,00 | |||
| 25,00 | |||
| 25,00 |
Реактивы
1. Раствор ЭДТА с молярной концентрацией ~ 0,05 моль/дм 3 .
2. Стандартный раствор Cu(II) с титром 2,00×10 -3 г/дм 3 .
3. Н 2 О дистиллированная.
4. Аммиачный буфер с рН~ 8 – 8,5.
5. Мурексид, индикаторная смесь.
Задачи
1. Вычислите α 4 для ЭДТА при pH=5, если константы ионизации ЭДТА следующие: K 1 =1,0·10 -2 , K 2 =2,1·10 -3 , K 3 =6,9·10 -7 , K 4 =5,5·10 -11 .
2. Постройте кривую титрования 25,00 мл 0,020 М раствора никеля 0,010 М раствором ЭДТА при pH=10, если константа устойчивости
К NiY = 10 18,62 . Вычислите p после добавления 0,00; 10,00; 25,00; 40,00; 50,00 и 55,00 мл титранта.
3. На титрование 50,00 мл раствора, содержащего ионы кальция
и магния, потребовалось 13,70 мл 0,12 М раствора ЭДТА при pH=12 и 29,60 мл при pH=10. Выразите концентрации кальция и магния в растворе в мг/мл.
4. При анализе в 1 л воды найдено 0,2173 г оксида кальция и 0,0927 г оксида магния. Вычислите, какой объём ЭДТА концентрации 0,0500 моль/л был затрачен на титрование.
5. На титрование 25,00 мл стандартного раствора, содержащего 0,3840 г сульфата магния, израсходовано 21,40 мл раствора трилона Б. Вычислите титр этого раствора по карбонату кальция и его молярную концентрацию.
6. На основании констант образования (устойчивости) комплексонатов металлов, приведенных ниже, оцените возможность комплексонометрического титрования ионов металлов при pH = 2; 5; 10; 12.
7. При титровании 0,01 М раствора Ca 2+ 0,01 М раствором ЭДТА при pH=10 константа устойчивости K CaY = 10 10,6 . Вычислите, какой должна быть условная константа устойчивости комплекса металла с индикатором при pH=10, если в конечной точке титрования =.
8. Константа кислотной ионизации индикатора, используемого при комплексонометрическом титровании, равна 4,8·10 -6 . Вычислите содержание кислотной и щелочной форм индикатора при pH = 4,9, если его общая концентрация в растворе составляет 8,0·10 -5 моль/л. Определите возможность использования данного индикатора при титровании раствора
с pH=4,9, если цвет его кислотной формы совпадает с цветом комплекса.
9. Для определения содержания алюминия в образце навеску образца 550 мг растворили и добавили 50,00 мл 0,05100 М раствора комплексона III. Избыток последнего оттитровали 14,40 мл 0,04800 М раствором цинка (II). Рассчитайте массовую долю алюминия в образце.
10. При разрушении комплекса, содержащего висмут и йодид-ионы, последние титруют раствором Ag(I), а висмут – комплексоном III.
Для титрования раствора, содержащего 550 мг образца, требуется 14,50 мл 0,05000 М раствора комплексона III, а на титрование йодид-иона, содержащегося в 440 мг образца, затрачивается 23,25 мл 0,1000 М раствора Ag(I). Рассчитайте координационное число висмута в комплексе, если йодид-ионы являются лигандом.
11.
Образец массой 0,3280 г, содержащий Pb, Zn, Cu, растворили
и перевели в мерную колбу на 500,00 см 3 . Определение вели в три этапа:
а) на титрование первой порции раствора объемом 10,00 см 3 , содержащего Pb, Zn, Cu, затрачено 37,50 см 3 0,0025 М раствора ЭДТА; б) во второй порции объемом 25,00 см 3 замаскировали Cu, а на титрование Pb и Zn израсходовано 27,60 см 3 ЭДТА; в) в третьей порции объемом 100,00 см 3 замаскировали Zn
и Cu, на титрование Pb затрачено 10,80 см 3 ЭДТА. Определите массовую долю Pb, Zn, Cu в образце.
Кривые титрования
В редоксметрии кривые титрования строят в координатах Е = f
(C R
),
они иллюстрируют графическое изменение потенциала системы в процессе титрования. До точки эквивалентности потенциал системы рассчитывается по отношению концентраций окисленной и восстановленной форм определяемого вещества (потому что до точки эквивалентности одна из форм титранта практически отсутствует), после точки эквивалентности – по отношению концентраций окисленной и восстановленной форм титранта (потому что после точки эквивалентности определяемое вещество оттитровано практически полностью).
Потенциал в точке эквивалентности определяется по формуле
 , (26)
, (26)
где – число электронов, участвующих в полуреакциях;
– стандартные электродные потенциалы полуреакций.
На рис. 10 представлена кривая титрования раствора щавелевой кислоты H 2 C 2 O 4 раствором перманганата калия KMnO 4 в кислой среде
( = 1 моль/дм 3).

Рис. 10. Кривая титрования 100,00 см 3 раствора щавелевой
кислоты H 2 C 2 O 4 с С 1/ z = 0,1000 моль/дм 3 раствором перманганата
калия KMnO 4 с С 1/ z = 0,1000 моль/дм 3 при =1 моль/дм 3
Потенциал полуреакции MnO 4 - + 5e + 8H + → Mn 2+ + 4H 2 O зависит от рН среды, так как в полуреакции участвуют ионы водорода.
Перманганатометрия
Титрантом является раствор перманганата калия KMnO 4 , являющийся сильным окислителем. Основное уравнение:
MnO 4 - +8H + + 5e = Mn 2+ + 4H 2 O, ![]() =+1,51 В.
=+1,51 В.
М 1/ z (KMnO 4)= ![]() г/моль.
г/моль.
В слабокислых, нейтральных и слабощелочных средах вследствие меньшего окислительно-восстановительного потенциала перманганат-ион восстанавливается до Mn +4 .
MnO 4 - +2H 2 O + 3e = MnО 2 ¯ + 4OH - , ![]() = +0,60 В.
= +0,60 В.
М 1/ z (KMnO 4)= 158,03/3= 52,68 г/моль.
В щелочной среде раствор перманганата калия восстанавливается
до Mn +6 .
MnO 4 - + 1e = MnO 4 2- , ![]() = +0,558 В.
= +0,558 В.
М 1/ z (KMnO 4)= 158,03 г/моль.
Для исключения побочных реакций титрование перманганатом калия проводят в кислой среде, которую создают серной кислотой. Соляную кислоту для создания среды применять не рекомендуется, так как перманганат калия способен окислять хлорид-ион.
2Cl - – 2e = Cl 2 , = +1,359 В.
Наиболее часто перманганат калия применяют в виде раствора
с молярной концентрацией эквивалента ~ 0,05 – 0,1 моль/дм 3 . Он не является первичным стандартом в силу того, что водные растворы перманганата калия способны окислять воду и органические примеси в ней:
4MnO 4- + 2H 2 O = 4MnО 2 ¯+ 3O 2 + 4OH -
Разложение растворов перманганата калия ускоряется в присутствии диоксида марганца. Поскольку диоксид марганца является продуктом разложения перманганата, этот осадок оказывает автокаталитический эффект на процесс разложения.
Твердый перманганат калия, применяемый для приготовления растворов, загрязнен диоксидом марганца, поэтому приготовить раствор из точной навески нельзя. Для того чтобы получить достаточно устойчивый раствор перманганата калия, его после растворения навески KMnO 4 в воде оставляют в темной бутыли на несколько дней (или кипятят), а затем отделяют MnO 2 ¯ фильтрованием через стеклянный фильтр (применять бумажный фильтр нельзя, так как он реагирует с перманганатом калия, образуя диоксид марганца).
Окраска раствора перманганата калия настолько интенсивна,
что индикатор в этом методе не требуется. Для того чтобы придать заметную розовую окраску 100 см 3 воды, достаточно 0,02 – 0,05 см 3 раствора KMnO 4
с молярной концентрацией эквивалента 0,1 моль/дм 3 (0,02 М). Окраска перманганата калия в конечной точке титрования неустойчивая и постепенно обесцвечивается в результате взаимодействия избытка перманганата
с ионами марганца (II), присутствующими в конечной точке в относительно большом количестве:
2MnO 4 - + 3Mn 2+ + 2H 2 O « 5MnО 2 ¯ + 4H +
Стандартизацию рабочего раствора KMnO 4 проводят по оксалату натрия или щавелевой кислоте (свежеперекристаллизованной и высушенной при 105°С).
Используют растворы первичных стандартов с молярной концентрацией эквивалента С (½ Na 2 C 2 O 4) = 0,1000 или 0,05000 моль/л.
C 2 O 4 2- – 2e ® 2CO 2 , = -0,49 В
Титриметрический анализ (объемный анализ) -- метод количественного анализа, основанный на измерении объема или массы реагента, требующегося для реакции с исследуемым веществом. Титриметрический анализ широко применяется в биохимических, клинических, санитарно-гигиенических и других лабораториях в экспериментальных исследованиях и для клинических анализов. Например, при установлении кислотно-щелочного равновесия, определении кислотности желудочного сока, кислотности и щелочности мочи и др. Титриметрический анализ служит также одним из основных методов химического анализа в контрольно-аналитических аптечных лабораториях.
Количество исследуемого вещества при титриметрическом анализе определяют путем титрования: к точно отмеренному объему раствора исследуемого вещества постепенно приливают раствор другого вещества известной концентрации до тех пор, пока его количество не станет химически эквивалентным количеству исследуемого вещества. Состояние эквивалентности называется точкой эквивалентности титрования. Применяемый для титрования раствор реактива известной концентрации называют титрованным раствором (стандартным раствором или титрантом): точная концентрация титрованного раствора может быть выражена титром (г/мл), нормальностью (экв/л) и др.
К реакциям, используемым при титриметрическом анализе, предъявляются следующие требования: вещества должны реагировать в строго количественных (стехиометрических) отношениях без побочных реакций, реакции должны протекать быстро и практически до конца; для установления точки эквивалентности необходимо применять достаточно надежные способы, влияние посторонних веществ на ход реакции должно быть исключено. Кроме того, желательно, чтобы при титриметрическом анализе реакции протекали при комнатной температуре.
Точку эквивалентности в титриметрическом анализе определяют по изменению окраски титруемого раствора или индикатора, вводимого в начале или в процессе титрования, изменению электропроводности раствора, изменению потенциала электрода, погруженного в титруемый раствор, изменению величины тока, оптической плотности и др.
Одним из широко применяемых способов фиксации точки эквивалентности является индикаторный метод. Индикаторы -- вещества, которые дают возможность установить конечную точку титрования (момент резкого изменения окраски титруемого раствора). Наиболее часто индикатор добавляют ко всему титруемому раствору (внутренний индикатор). При работе с внешними индикаторами периодически берут каплю титруемого раствора и смешивают с каплей раствора индикатора или помещают на индикаторную бумагу (что приводит к потерям анализируемого вещества).
Процесс титрования изображают графически в виде кривых титрования, которые позволяют наглядно представить весь ход титрования и выбрать индикатор, наиболее пригодный для получения точных результатов, т.к. кривую титрования можно сопоставить с интервалом изменения окраски индикатора.
Ошибки в титриметрическом анализе могут быть методическими и специфическими, обусловленными особенностями данной реакции. Методические ошибки связаны с особенностями метода титрования и зависят от погрешностей измерительных приборов, калибровки мерной посуды, пипеток, бюреток, неполного отекания жидкостей по стенкам мерной посуды.
Специфические ошибки обусловлены особенностями данной реакции и зависят от константы равновесия реакции и от точности обнаружения точки эквивалентности. фармацевтический лекарство молекула анальгин
Методы титриметрического анализа в зависимости от реакций, лежащих в их основе, подразделяются на следующие основные группы:
- 1. Методы нейтрализации, или кислотно-основного титрования, основаны на реакциях нейтрализации, т. е. на взаимодействии кислот и оснований. Эти методы включают ацидиметрию (количественное определение оснований с помощью титрованных растворов кислот), алкалиметрию (определение кислот с помощью титрованных растворов оснований), галометрию (количественное определение солей с помощью оснований или кислот, если они реагируют с солями в стехиометрических соотношениях).
- 2. Методы осаждения основаны на титровании веществ, образующих в определенной среде нерастворимые соединения, например, соли бария, серебра, свинца, цинка, кадмия, ртути (II), меди (III) и др. К этим методам относят аргентометрию (титрование раствором нитрата серебра), меркурометрию (титрование раствором нитрата закисной ртути) и др.
- 3. Методы комплексообразования, или комплексометрия (меркуриметрия, фторометрия и др.), основаны на применении реакций, при которых образуются комплексные соединения, например Ag+ + 2CN- Ы Ag (CN)2]. Методы комплексообразования тесно связаны с методами осаждения, т.к. многие реакции осаждения сопровождаются комплексообразованием, а образование комплексов -- выпадением в осадок малорастворимых соединений.
- 4. Методы окисления -- восстановления, или оксидиметрия, включают перманганатометрию, хроматометрию (бихроматометрию), йодометрию, броматометрию, цериметрию, ванадометрию и др.
Заполненной титрантом до нулевой отметки. Титровать, начиная от других отметок, не рекомендуется, так как шкала бюретки может быть неравномерной. Заполнение бюреток рабочим раствором производят через воронку или с помощью специальных приспособлений, если бюретка полуавтоматическая. Конечную точку титрования (точку эквивалентности) определяют индикаторами или физико-химическими методами (по электропроводности, светопропусканию, потенциалу индикаторного электрода и т. д.). По количеству пошедшего на титрование рабочего раствора рассчитывают результаты анализа.
Виды титриметрического анализа
Титриметрический анализ может быть основан на различных типах химических реакций:
- кислотно-основное титрование - реакции нейтрализации ;
- окислительно-восстановительное титрование (перманганатометрия, иодометрия , хроматометрия) - окислительно-восстановительные реакции ;
- осадительное титрование (аргентометрия) - реакции, протекающие с образованием малорастворимого соединения, при этом изменяются концентрации осаждаемых ионов в растворе;
- комплексонометрическое титрование - реакции, основанные на образовании прочных комплексных соединений ионов металлов с комплексоном (обычно ЭДТА), при этом изменяются концентрации ионов металлов в титруемом растворе.
Типы титрования
Различают прямое, обратное титрование и титрование заместителя.
- При прямом титровании к раствору определяемого вещества (аликвоте или навеске, титруемому веществу) добавляют небольшими порциями раствор титранта (рабочий раствор).
- При обратном титровании к раствору определяемого вещества добавляют сначала заведомый избыток специального реагента и затем титруют его остаток, не вступивший в реакцию.
- При заместительном титровании к раствору определяемого вещества добавляют сначала заведомый избыток специального реагента и затем титруют один из продуктов реакции между анализируемым веществом и добавленным реагентом.
См. также
Ссылки
Wikimedia Foundation . 2010 .